HISTIOCYTIC DISORDERS
HISTIOCYTIC DISORDERS
The histiocytoses are a group of heterogeneous reactive and neoplastic disorders in which cells of the dendritic or monocyte–macrophage lineage proliferate in various tissues, including the skin. They have been the source of considerable confusion due to their relative rarity, the use of imprecise terminology, and a lack of knowledge of the precise lineage of lesional cells.
The term ‘histiocyte’ denotes a group of tissue-based immune cells that includes both macrophages and dendritic cells. Macrophages display strong phagocytic capabilities and function predominantly as antigen presenting cells, whereas dendritic cells are primarily accessory cells with antigen presenting functions.1,2 Distinction between these two groups is often not clear since both cell types display considerable plasticity, their characteristics changing according to their stage of development and/ or the influence of the microenvironment in which they are present. This results in considerable functional, morphological, and immunophenotypic overlap.1–3
molecular, clinical, and imaging characteristics. It recognized five groups of diseases:
• cutaneous and mucocutaneous (non-Langerhans cell) histiocytoses,
• Rosai-Dorfman disease (RDD),
• malignant histiocytoses,
• hemophagocytic lymphohistiocytosis (HLH) and macrophage activation syndrome. Historically, LCH has been separated from non-Langerhans cell histiocytoses.1,6 However, the discovery of shared genetic abnormalities and clinical features between LCH and Erdheim-Chester disease (ECD) has led to both being grouped within the Langerhans cell-related group, together with indeterminate cell histiocytosis. Cutaneous and mucocutaneous histiocytoses encompass the xanthogranuloma family of diseases as well as multicentric reticulohistiocytosis (MRH). Cutaneous RDD is also included in this group, separated from classical sporadic RDD and RDD secondary to predisposing inherited conditions. Malignant histiocytosis includes cases previously diagnosed as histiocytic sarcoma (HS), interdigitating dendritic cell (IDC) sarcoma, Langerhans cell sarcoma (LCS), and indeterminate cell sarcoma. These may arise de novo or secondary to other malignancies. Lastly, there is a category of HLH and macrophage activation syndrome, which may be related to an inherited genetic disorder or secondary to other etiologies. These disorders are detailed in Table 29.5.
The majority of histiocytes originate from a CD34+ bone marrow progenitor, although some may derive from mesenchymal cells (e.g., FDCs) or even lymphocytes. A simplistic view of subsequent development holds that one of two pathways is followed with maturation into either CD14– or CD14+ cells. The former give rise to Langerhans cells while the latter are the source of dermal dendritic cells or monocyte/macrophages.4
The system used to define the various entities in this edition is based on the recent revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineage recently proposed by the Histiocyte Society.5 This classification takes cognisance of histologic, phenotypic,
Two additional entities not listed in the table are discussed first, these are cutaneous Kikuchi-Fujimoto disease (KFD) and intralymphatic histiocytosis (IH).
Langerhans cell family of histiocytoses Langerhans cell histiocytosis Congenital self-healing histiocytosis Indeterminate dendritic cell tumor Erdheim-Chester disease
Cutaneous and mucocutaneous histiocytoses (non-Langerhans cell) Xanthogranuloma family
1489 Intralymphatic histiocytosis
Differential diagnosis
The differential diagnosis includes pityriasis lichenoides, lupus erythematosus, and the adverse antibiotic-induced eruptions associated with EBV infection.14,25 In pityriasis lichenoides, there are necrotic keratinocytes and hydropic degeneration of basal cells. However, the infiltrate is predominantly lymphocytic, and nuclear debris, if present, are very focal. Distinction from lupus erythematosus is extremely difficult, particularly as the lymph node changes may be identical. Distinction is based on clinicopathological correlation, immunofluorescence, and the scarcity of histiocytes and presence of plasma cells in cutaneous lesions of lupus erythematosus. The histologic features of the adverse antibiotic-induced eruptions associated with EBV infection are identical to those seen in KFD, and distinction is based on the clinical information.
• Juvenile xanthogranuloma
• Benign cephalic histiocytosis
• Generalized eruptive histiocytosis
• Xanthoma disseminatum
• (scalloped cell granuloma)
• Papular xanthoma
• Progressive nodular histiocytosis
• (spindle cell xanthogranuloma) (Cutaneous Rosai-Dorfman disease) Multicentric reticulohistiocytosis
Malignant histiocytoses Langerhans cell sarcoma Follicular dendritic cell sarcoma Interdigitating dendritic cell sarcoma Histiocytic sarcoma
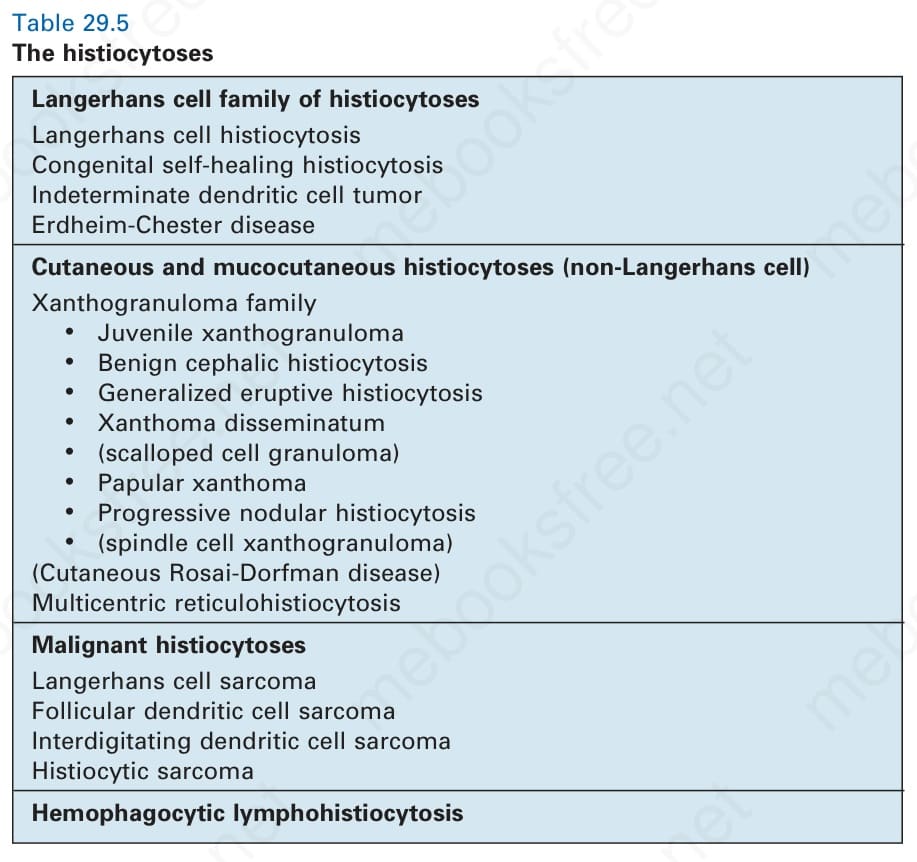
Table 29.5 The histiocytoses