Plasmablastic lymphoma
Plasmablastic lymphoma
Clinical features Plasmablastic lymphoma (PL) is a rare neoplasm of blastic cells displaying morphological and immunophenotypic features of terminal B-cell differentiation. Although originally described in the oral mucosa in HIV-positive individuals, and referred to as plasmablastic lymphoma of the oral mucosa in the 2001 WHO lymphoma classification, a broader spectrum of sites and clinical presentations is now appreciated.1–3
PL is seen most frequently in HIV-positive individuals, but is now also described following organ transplantation and in apparently immune competent individuals.1,4–11 It has also been rarely documented in association with non-transplant-related iatrogenic immunosuppression.12–14 A wide age range may be affected, including pediatric patients.5 In the HIV-positive group the median age at presentation is 42 years, whilst HIV-negative and post-transplant patients tend to be older.4,5 Males are more frequently affected than females in all groups. The vast majority of cases involve extranodal sites and are advanced stage (III/IV) at presentation.4,5 The oral cavity is most frequently involved followed by the gastrointestinal tract and skin. Other sites include the anus, sinonasal cavities, orbit, bone, soft tissues, lung, and genitourinary system.4–9,15,16 Lymph nodes are only rarely involved as the presenting site or as part of disease dissemination.4–8
Most patients present with masses.1,3,7,8 Cutaneous involvement usually manifests as solitary or grouped, purple or erythematous nodules, or more rarely plaques on the extremities.7,15,17–19 Most patients die within a year of diagnosis,1,4,5 although the outlook may be less bleak in patients undergoing treatment with antiviral therapies.10,20
Pathogenesis and histologic features Immunodeficiency predisposes to the development of plasmablastic lymphoma. As mentioned above, this may be due to HIV infection or a consequence of iatrogenic immunosuppression, usually in the post-transplant setting.4,5 Patients with no obvious history of immunosuppression tend to be aged >50 years, and therefore may be intrinsically more prone to EBV-related lymphoproliferative disorders due to immunological deterioration or senescence.4–6 EBV is present in neoplastic lymphocytes in the majority of cases and shows a type I latency, implying a significant role in the pathogenesis of at least some tumors.1,4–8,21,22 Translocations involving the MYC gene are found in 50–67% of cases, suggesting a role for this oncogene in the majority of tumors.4,5,12
Histologically, plasmablastic lymphoma is characterized by sheets of blast cells with features of plasmablasts (Figs 29.241 and 29.242). These typically
1478 Cutaneous lymphoproliferative diseases and related disorders
possess round, eccentrically placed nuclei with moderately clumped chromatin and a single central nucleolus, or several smaller peripherally placed nucleoli. There is moderate to abundant basophilic cytoplasm, often with a perinuclear hof. The mitotic index is high and apoptotic cells are frequent. In the oral cavity and nasal and paranasal areas, these blast cells predominate, giving a monomorphic appearance to the infiltrate.1,3,4–8 However, at other extranodal sites and in lymph nodes, the cytological spectrum of tumor cells is broader and includes a larger proportion of cells showing greater degrees of plasmacytic differentiation.1,4–8,21
Immunohistochemically, tumor cells display a plasma cell phenotype with strong expression of plasma cell markers including CD138, CD79a, CD38, Vs38c, IRF4/MUM1, and BLIMP-1.5,23 They should be negative for CD19, CD20, and pax5. CD45 may be expressed weakly, and CD10 and CD56 may also be positive, particularly in post-transplant patients.4,5 Cytoplasmic immunoglobulins can be demonstrated in about 50% of cases (usually IgG and only very occasionally IgM or IgA) with either kappa or lambda light chain.1,7,8,23 T-cell antigens such as CD2 and CD4 are occasionally aberrantly expressed.5 The Ki-67 index is typically greater than 90%. EBV is detectable in up to 75% of cases using in situ hybridization for EBERs, but staining for LMP1 and EBNA2 is usually negative.1,4,5,7,8,22,24 Despite one report to the contrary, HHV8 should not be present in the neoplastic lymphocytes.1,4,5,8,23
Immunoglobulin heavy chain genes are clonally rearranged, and somatic hypermutation is seen in a proportion of cases.24
Differential diagnosis The differential diagnosis of PL includes other blastic lymphomas showing plasmablastic or plasmacytic differentiation. These include plasmablastic and anaplastic variants of plasma cell myeloma and plasmacytoma, diffuse large B-cell lymphoma with plasmacytic differentiation, Burkitt lymphoma with plasmacytic differentiation, ALK-positive large B-cell lymphoma, primary effusion lymphoma, and large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease (CD).
Plasmablastic and anaplastic variants of myeloma/plasmacytoma share many morphological and immunophenotypic features with PL.23 The presence of significant bone involvement, a monoclonal serum paraprotein, lower proliferation index, and negative staining for EBV favor a diagnosis of plasma cell myeloma. Diffuse large B-cell lymphoma with plasmacytic differentiation is usually strongly positive for CD20 and CD45, and these markers – together with coexpression of CD10 and bcl-6 – should also allow reliable discrimination of Burkitt lymphoma. ALK-positive large B-cell lymphoma is also morphologically similar to PL, may lack CD20 and CD45, and express plasma cell markers. However, such cases are always positive for ALK protein, with a granular cytoplasmic staining pattern, usually express cytoplasmic IgA, and often coexpress CD4 and CD57.25
The neoplastic lymphocytes in primary effusion lymphomas also lack B-cell markers, react with antibodies to plasma cell antigens including CD138 and Vs38c, and are frequently EBV positive. However, they tend to be larger and more pleomorphic than those seen in PL, and uniformly show nuclear positivity with antibodies to HHV8. In addition, location within body cavities strongly supports a diagnosis of primary effusion lymphoma, and while extracavitary examples of the tumor exist, they more frequently express B-cell antigens.26 Large B-cell lymphomas arising in HHV8-associated multicentric CD were originally also referred to as PLs. However, they more frequently express CD20, are usually negative for EBV, and show cytoplasmic IgM lambda restriction. A background of plasma cell CD and stippled nuclear staining for the HHV8 also support this diagnosis over that of PL.
including full, thorough clinical examination, computerized tomography of trunk, and bone marrow examination, are mandatory for patients presenting with B-cell lymphoma presenting in the skin. The results of these studies should alert the pathologist to the possibility of underlying nodal disease, and may be the only reliable means of differentiating DLBCL from primary cutaneous diffuse large B-cell lymphoma, leg type, and large cell variants of PCFCL.
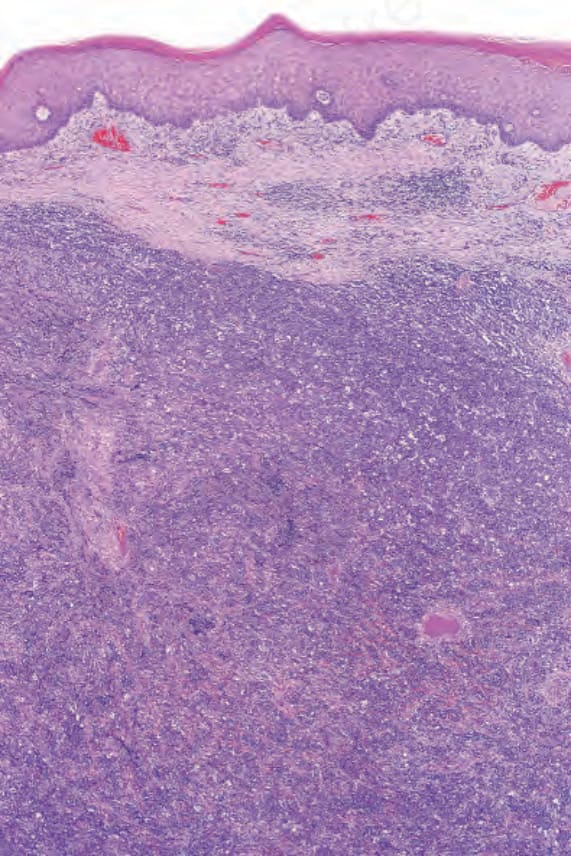
Fig. 29.241 Plasmablastic lymphoma: there is a dense dermal tumor cell infiltrate.
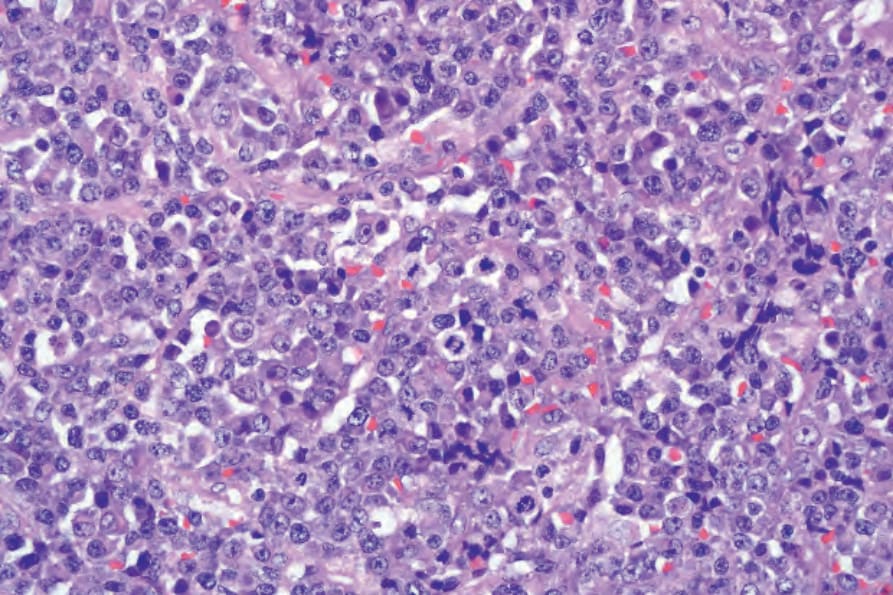
Fig. 29.242 Plasmablastic lymphoma: high-power view of plasmablasts with central nucleoli and occasional mature plasma cells.
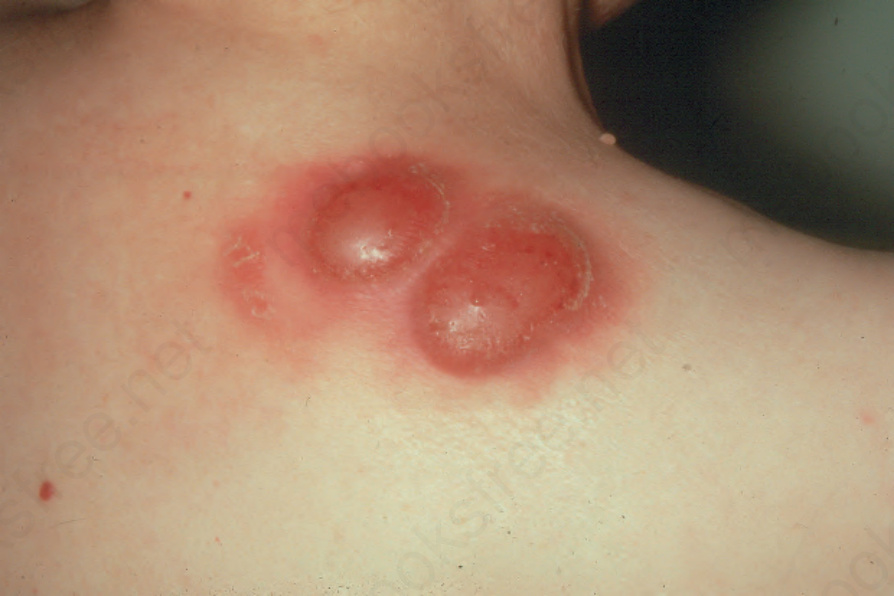
Fig. 29.243 Nodal B-cell lymphoma: skin involvement has presented with large tumor nodules showing surrounding erythema. By courtesy of R.A. Marsden, St George’s Hospital, London, UK.
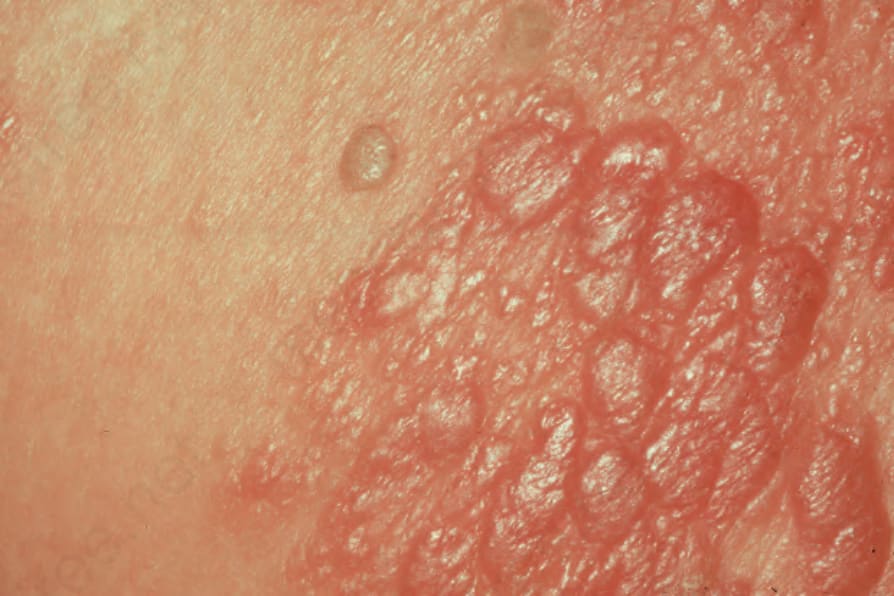
Fig. 29.244 Primary nodal B-cell lymphoma: there are numerous confluent erythematous papules. By courtesy of R.A. Marsden, St George’s Hospital, London, UK