Lymphomatoid papulosis
Lymphomatoid papulosis
Clinical features First described in 1968, lymphomatoid papulosis is a chronic, self-healing eruption.1 It is now classified as an indolent lymphoproliferative disorder in the WHO classification.
1434 Cutaneous lymphoproliferative diseases and related disorders
presentation in Japan.18–21 Pediatric and adolescent disease constitutes 6–10% of cases.3,4 The clinical presentation may be alarming, with the rapid development of large ulcerating lesions in addition to the usual papular eruption. However, the disease is otherwise typical.4,22–24
Lymphomatoid papulosis may run a short self-limiting course, or a more protracted course of 5–10 years or longer. Whatever the scenario, the outcome is generally benign, with treatment only being required when lesions are particularly numerous and/or cosmetically disturbing.4 However, between 9% and 19% of cases are associated with another lymphoma, such as mycosis fungoides, primary cutaneous anaplastic large cell lymphoma, or HL.3,25–29 The lymphoma may precede, arise concurrently with, or succeed lymphomatoid papulosis. The occurrence of lymphomatoid papulosis lesions restricted to the site of mycosis fungoides lesions has been described as persistent agmination of lymphomatoid papulosis.30,31
Lymphomatoid papulosis occurs more frequently in males (2 : 1). Patients are usually in their fifth decade, although a wide age range, including children, may be affected.2–8 The typical presentation is crops of erythematous papules, 0.5–1.0 cm across, which develop over the course of 3–4 weeks, become hemorrhagic and necrotic, and then heal, forming atrophic scars (Figs 29.102–29.104). The clinical features often overlap with those of pityriasis lichenoides acuta. Other lesions may be larger and nodular, and heal with deep varioliform scars. The condition commonly presents on the trunk and limbs, but occasionally other sites are involved. Numbers of lesions are variable, ranging from several to hundreds and take from a few weeks to several months to regress.9
Pathogenesis and histologic features The pathogenesis remains undetermined. Most cases appear to represent clonal proliferations of T lymphocytes, more likely a T-regulatory cell subset.3,28,32–35 Some studies have shown a high rate of apoptosis probably contributing to regression.36,37 This may be mediated by death-receptor pathway signaling via cell surface Fas (CD95) signaling and/or due to increased levels of the proapoptotic protein bax.3,38–41 An autocrine or paracrine growth control mechanism has been proposed, mediated by TGF-β secretion and signaling. Mutations of TGF-β signaling receptor genes results in disease progression.34,42,43 More recently, a distinctive subset of lymphomatoid papulosis cases characterized by predominantly localized lesions presenting in elderly patients and associated with chromosomal rearrangements involving 6p25.3 has been described.44
Rare clinical variants have been described. In regional lymphomatoid papulosis, lesions are limited to one body region for years, and seem to be more common in children.10–13 Mucosal involvement is exceptional.14–17 Oral lesions present as recurring and spontaneously regressing painful ulcers, nodules, or erythematous indurated plaques.15,17 Follicular and pustular variants have been described, as has a hydroa vacciniforme (HV)-like
In cases of lymphomatoid papulosis associated with another lymphoma, a common clonal identity can often be demonstrated, suggesting a common stem cell giving rise to both.34,45–50 This theory, supported by cytogenetic findings, holds that different tumor cell phenotypes with distinct histopathologies and behaviors arise from accumulated genetic alterations in subclones of a common, occult stem cell.34,45–50
Patients with lymphomatoid papulosis exhibit a variety of pathologies. An awareness of this spectrum is important as many of the patterns encountered can be mistaken for more aggressive lymphomas. The different types described overlap and may be seen in different biopsies from the same patient. Traditionally, the different patterns of lymphomatoid papulosis have been designated subtypes with a letter of the alphabet appended for identification. This is a rather arbitrary subdivision but is adhered to herein to highlight the range of specific features that may be encountered.51
1435 Primary cutaneous CD30-positive T-cell lymphoproliferative disorders
• Type A pattern (75–80% of cases) consists of a mixed, wedge-shaped dermal and rarely focally subcutaneous infiltrate containing large anaplastic cells (15–30 µm in diameter) with pleomorphic vesicular nuclei containing prominent nucleoli and abundant cytoplasm (Figs 29.105–29.109). These may be multinucleate and can resemble Reed-Sternberg cells. Mitotic figures are frequent (Fig. 29.110). In established lesions, these cells are scattered or arranged in small clusters, and admixed with neutrophils, eosinophils, plasma cells, lymphocytes, and histiocytes. Epidermotropism of large atypical cells is rare.3
• Type B pattern (5–10% of cases) has a preponderance of small to medium-sized lymphocytes with pleomorphic irregular and enlarged nuclei (Figs 29.111 and 29.112). The infiltrate displays a bandlike distribution in the upper dermis. Epidermotropism is prominent, and it simulates plaque stage mycosis fungoides, although Pautrier microabscesses, halos, and basal lymphocytic palisades are usually absent.9
• In type C pattern (7–10% of cases), the infiltrate is nodular with large clusters or cohesive sheets of type A cells with relatively few inflammatory cells. The features may be identical to those seen in
1436 Cutaneous lymphoproliferative diseases and related disorders
primary cutaneous anaplastic large cell lymphoma, and the two processes are only distinguished on the basis of clinical features.4
• Type D lymphomatoid papulosis is associated with prominent epidermal hyperplasia and marked epidermotropism. The intraepidermal lymphocytes are usually larger than those seen in type B lesions and express CD8 rather than CD4. However, large anaplastic cells, as seen in types A and C lymphomatoid papulosis, are usually absent.52
• So-called type E lymphomatoid papulosis is characterized by an angioinvasive growth pattern with destruction of blood vessel walls. The infiltrating lymphocytes may be small, medium, or large and are admixed with eosinophils and neotrophils. There is often associated necrosis. Clinically, these patients are slightly different from those with classical lymphomatoid papulosis, presenting with relatively few papulonodules that evolve into large flat ulcerations (eschar-like) measuring up to 4 cm in diameter.53
• A type F lymphomatoid papulosis has also been proposed for cases exhibiting prominent folliculotropism, although this is not an uncommon feature seen in association with other patterns of infiltration.3,54,55 Secondary changes, including follicular mucinosis, hyperplasia of hair follicle epithelium, folliculitis, and granulomatous inflammation, often ensue.3,55
• Recently, a specific subtype of lymphomatoid papulosis associated with a specific genetic abnormality has been described in a small number of patients.56 All have translocations involving the DUSP22-IRF4 locus at 6p25.3. These patients present with one to several eruptive papulonodular lesions limited to a single body area. Biopsy shows a cohesive nodular dermal infiltrate of medium to large blast cells. The overlying epidermis shows extensive colonization by small to medium lymphocytes, often with cerebriform nuclear contours. Pautrier microabscess-like collections may be seen, and the epidermal changes often resemble those seen in pagetoid reticulosis.56
It should be noted that not all cases of lymphomatoid papulosis neatly subclassify into the variants described above. In up to 10% of cases, there are overlapping features of two or more subtypes within a single lesion, and lesions displaying different patterns may be present at different sites and/or times within a single patient.3,57,58
Other features are common to all of the subtypes. Dermal edema and hemorrhage are often conspicuous. Some vessels may show fibrin deposition and occlusion. Reactive epidermal changes are variable, depending on the stage of evolution of the papule. Early lesions show intercellular edema and occasional intraepidermal lymphocytes. Intermediate lesions are characterized by variable necrosis of keratinocytes, intercellular edema, and intraepidermal cells, many of which are atypical. Intraepidermal polymorphs and erythrocytes are commonly found. Late lesions are characterized by extensive epidermal necrosis, ulceration, and the formation of a scaly, parakeratotic crust. Occasionally, there is striking pseudoepitheliomatous hyperplasia such that misdiagnosis as a squamous cell carcinoma or keratoacanthoma may result.59
Rare histologic patterns described include a myxoid variant with a sarcoma-like appearance and a syringotropic example.3,60,61
In all histologic patterns, the atypical lymphoid cells express CLA and are usually CD4+ and CD8–, although occasionally CD4–/CD8+ (type D and type E in particular), CD4–/CD8– and CD4+/CD8+ variants are encountered.3,52,53,62 Cytotoxic molecules, such as TIA-1, perforin, and granzyme B, are usually identifiable, irrespective of the CD4/CD8 status.3,63 There is variable expression of the pan-T-cell antigens, CD2, CD3, CD5, and CD7.4 The anaplastic cells in the types A and C variants express CD45 and CD30, but not CD15, or p80/ALK1 reactivity with CD25 may also be seen (Fig. 29.113).45,64–69 Although epithelial membrane antigen (EMA) is usually negative, it may be focally positive in some cases. Conversely, in type B lesions, CD30 is often, although not always, negative.3,33 Although earlier studies suggested an absence of CD56, more recently expression of this molecule has frequently been seen.3,68,69 In general, tumors of natural killer (NK)/T-cell origin have been generally associated with a very poor prognosis, but this does not appear to be the case in lymphomatoid papulosis. Most cases (75–80%) of lymphomatoid papulosis are also positive for IRF4 (MUM1), but this protein is also expressed in most primary cutaneous
1437 Primary cutaneous CD30-positive T-cell lymphoproliferative disorders
anaplastic large cell lymphomas and is not a useful marker for distinguishing between these two ends of a spectrum.70,71 Some cases of lymphomatoid papulosis type D express TCR-γ.72
TCR gene rearrangements are detected in approximately 60% of cases.3,26,32,33,44
Differential diagnosis Expression of CD30 is not a reliable discriminator for differentiating cutaneous CD30+ T-cell lymphoproliferative disorders from reactive inflammatory skin conditions or other types of lymphoma. An increasing number of infectious skin diseases have been shown to contain significant numbers of CD30+ cells and mimic lymphomatoid papulosis. These include cutaneous lesions in various viral infections, including herpes virus, molluscum contagiosum, parapox virus (milker’s nodule), EBV, HTLV-1, and HIV.67,70,73–78 Lesions of scabies, syphilis, and superficial fungal infections may also contain CD30+ cells.66,79,80 Noninfectious cutaneous inflammatory processes, such as pityriais lichenoides et varioliformis acuta, atopic dermatitis, and drug reactions (particularly to anticonvulsants), may also harbor small numbers of CD30+ cells.81–84 Cutaneous eruption of lymphocyte recovery may also contain CD30+ cells.66,85
In most of these disorders, the CD30+ cells lack the anaplastic features and are present in fewer numbers than in lymphomatoid papulosis. They also tend to be scattered, rather than in small clusters, but this is not always the case.66 However, close clinicopathological correlation remains essential.
Type B and type D lymphomatoid papulosis displaying epidermotropism may be impossible to distinguish from other epidermotropic lymphomas such as mycosis fungoides (including pagetoid reticulosis) and primary cutaneous CD8-positive aggressive epidermotropic T-cell lymphoma on the basis of pathological features. In such situations, the diagnosis is dependent on the clinical features.
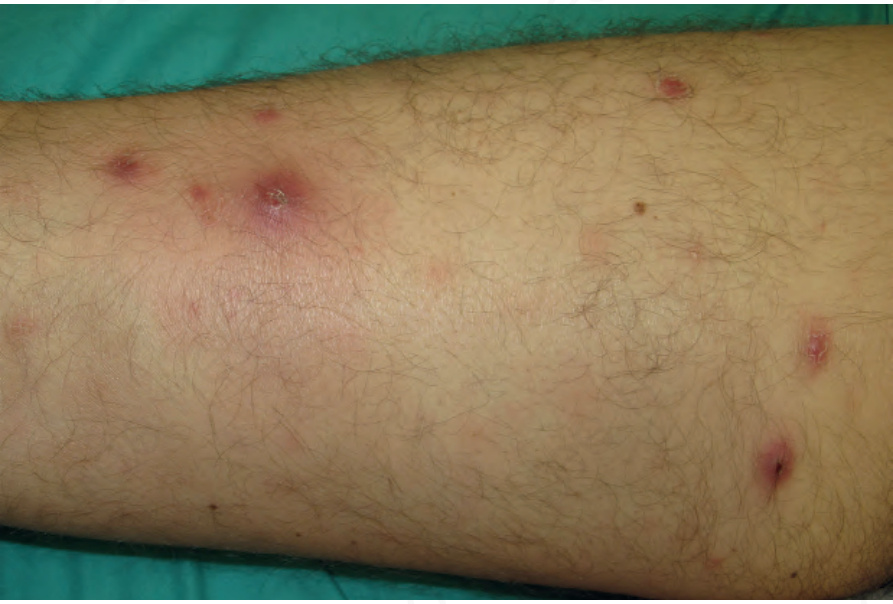
Fig. 29.102 Lymphomatoid papulosis: multiple variably sized papules on a limb. Courtesy of Dr Teresa Estrach Panella, Barcelona, Spain.
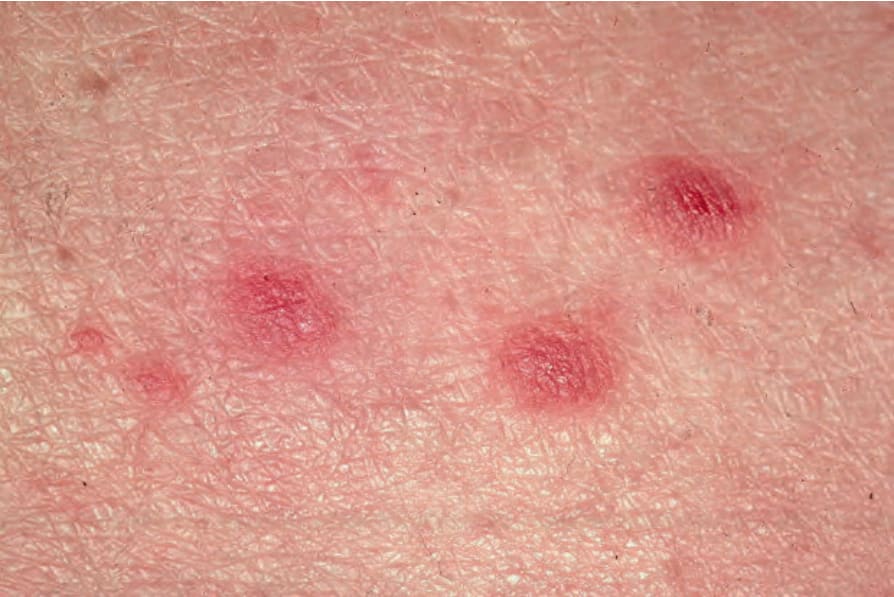
Fig. 29.103 Lymphomatoid papulosis: close-up view of erythematous papules. By courtesy of R.A. Johnson, MD, Massachusetts General Hospital, Harvard Medical School, Boston, USA.
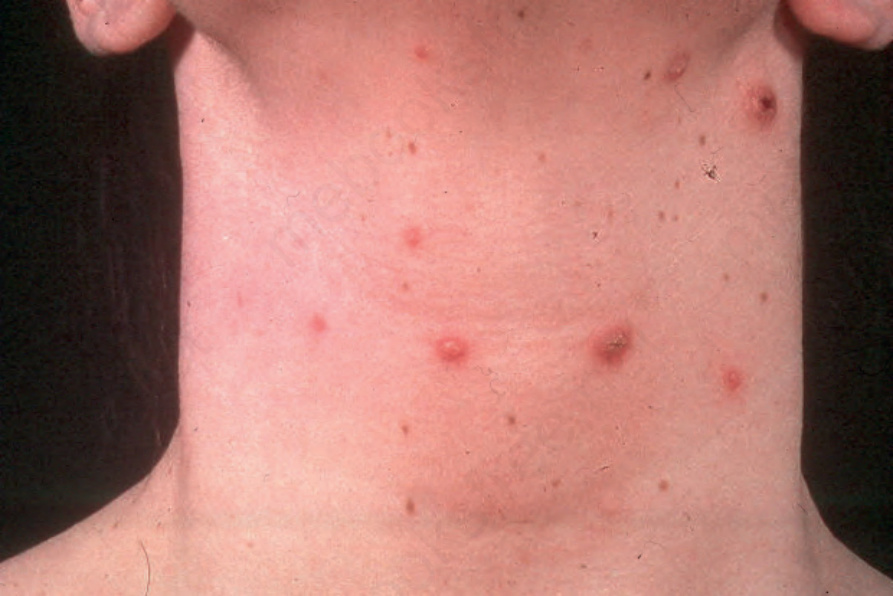
Fig. 29.104 Lymphomatoid papulosis: in this example, lesions are present on the neck. By courtesy of the Institute of Dermatology, London, UK.
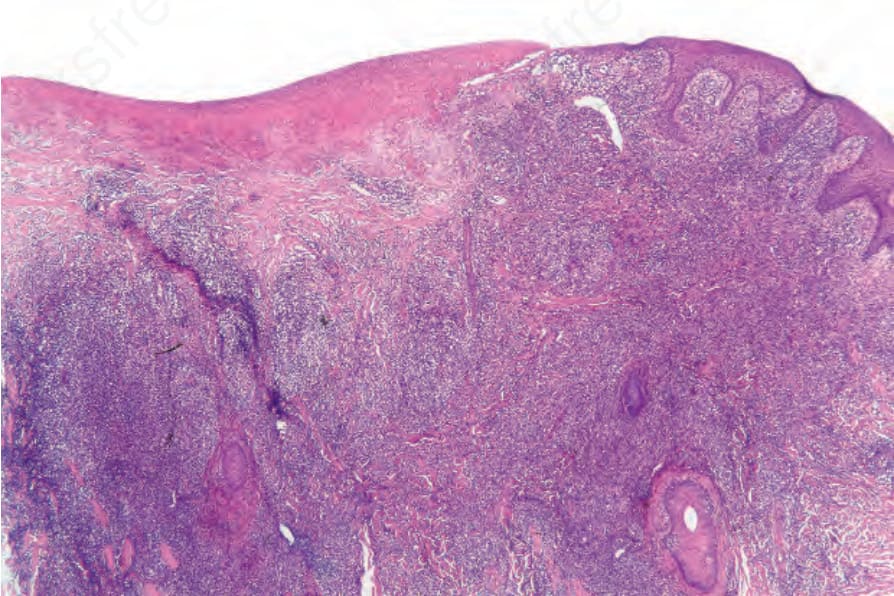
Fig. 29.105 Lymphomatoid papulosis: low-power view showing ulceration and a dense dermal infiltrate.
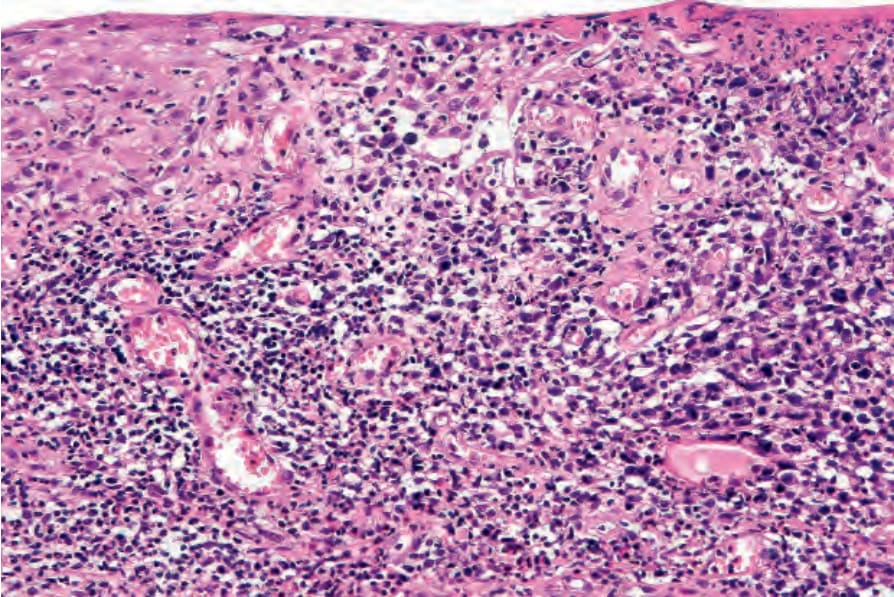
Fig. 29.106 Lymphomatoid papulosis: the epidermis is infiltrated by atypical pleomorphic lymphocytes.
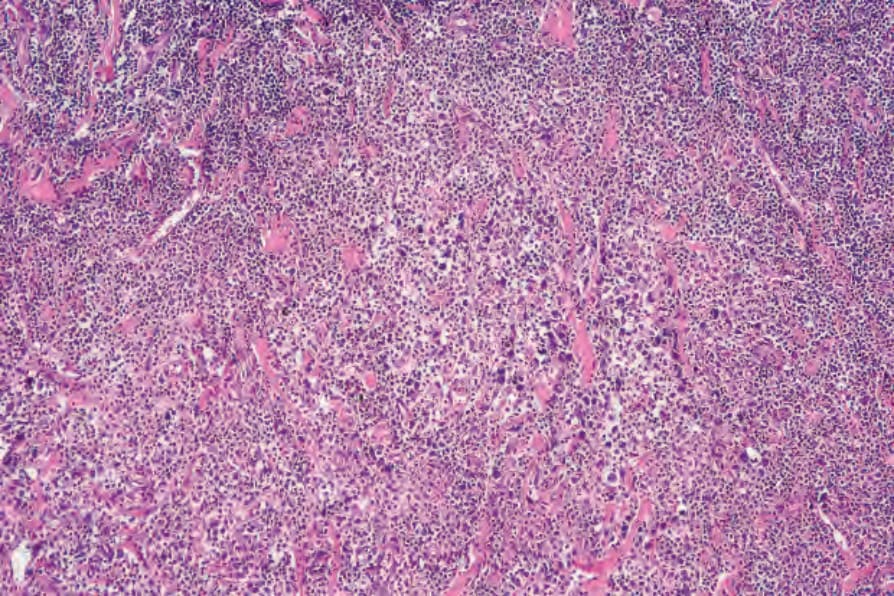
Fig. 29.107 Lymphomatoid papulosis: dense dermal infiltrate. Even at this magnification, the cytological atypia is obvious.
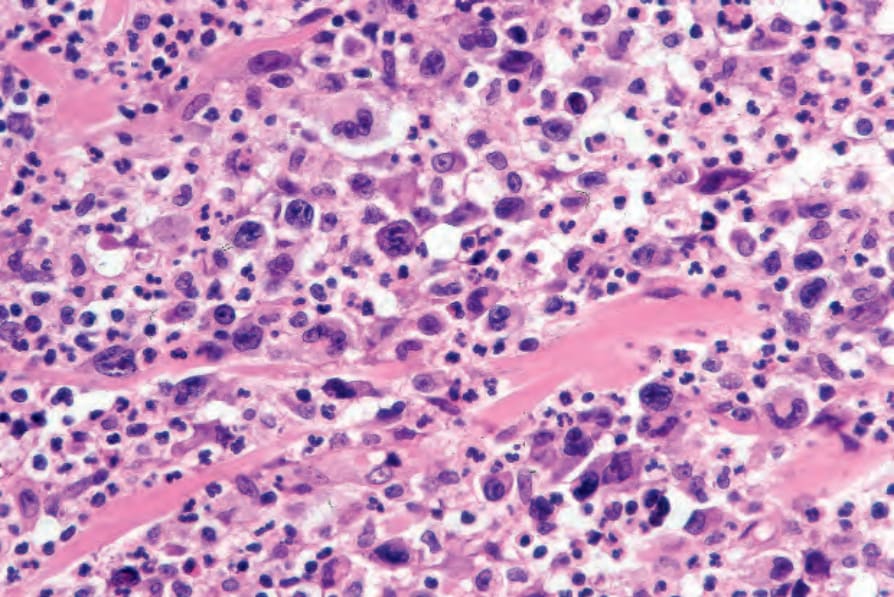
Fig. 29.108 Lymphomatoid papulosis: the lymphocytes have highly irregular hyperchromatic or vesicular nuclei. Note the background population of neutrophils.
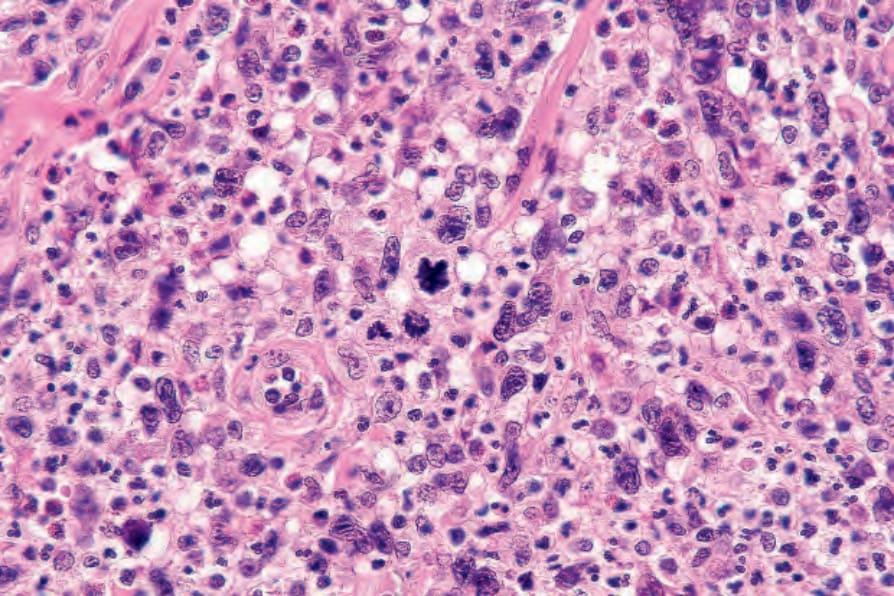
Fig. 29.109 Lymphomatoid papulosis: multiple mitotic figures are present.
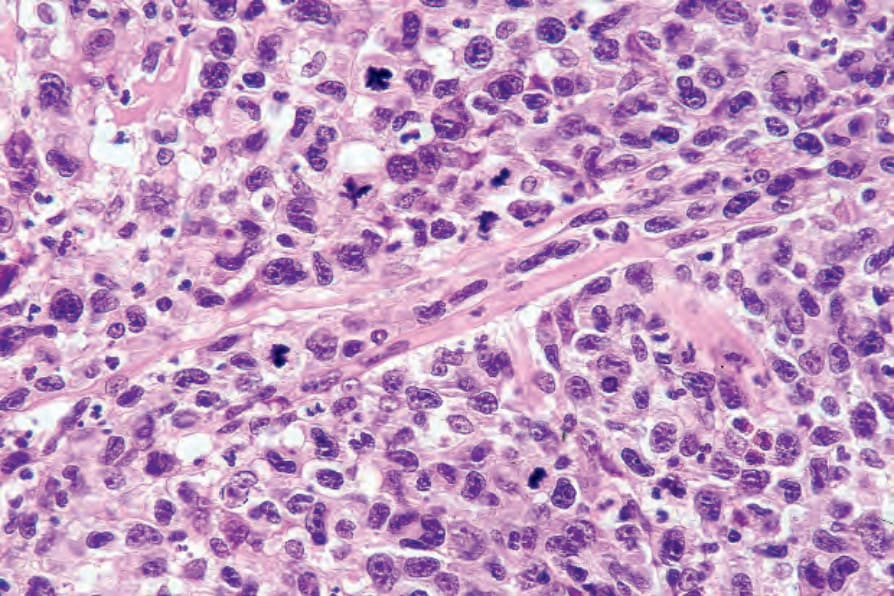
Fig. 29.110 Lymphomatoid papulosis: an atypical mitosis is seen just above the center of the field.
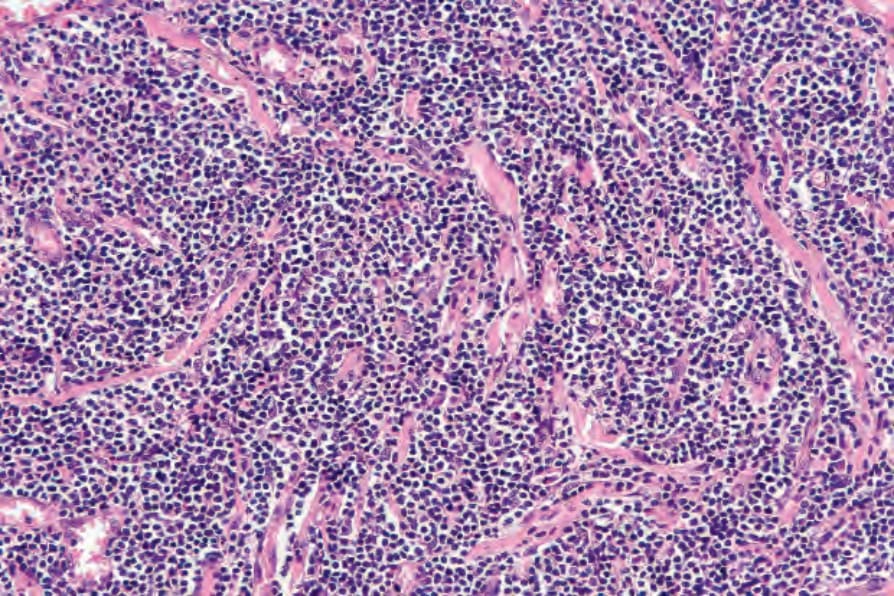
Fig. 29.111 Lymphomatoid papulosis: there is a dense infiltrate of type B cells.
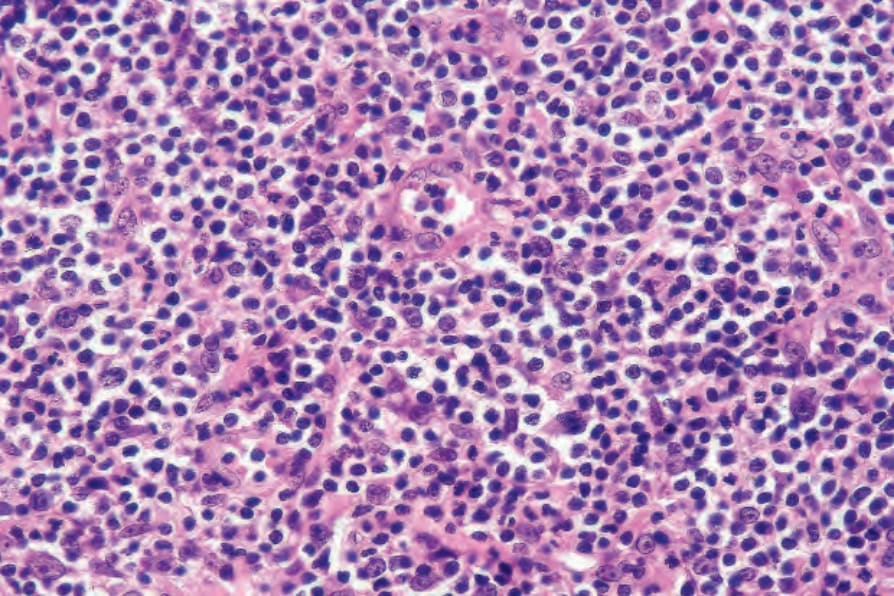
Fig. 29.112 Lymphomatoid papulosis: the type B cells have enlarged, irregular hyperchromatic nuclei and scanty cytoplasm reminiscent of mycosis cells.
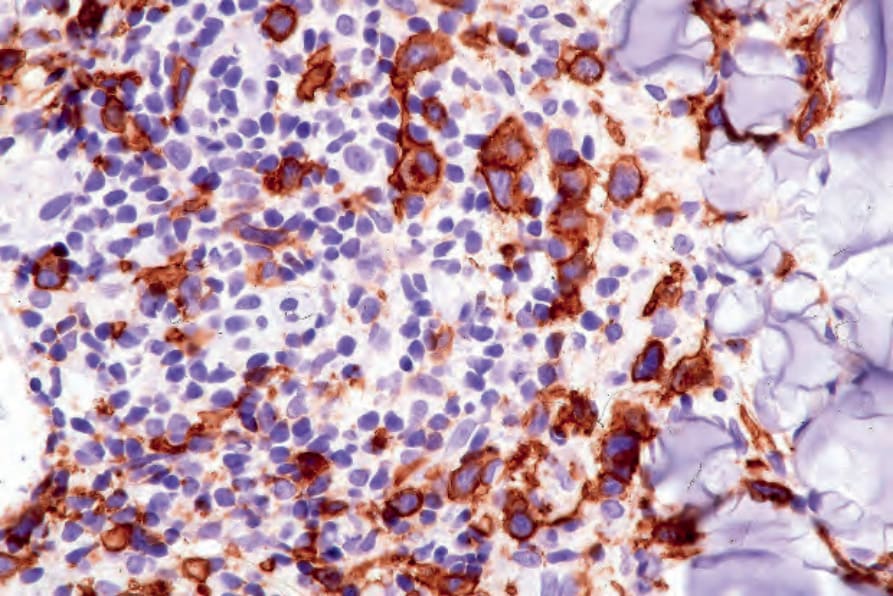
Fig. 29.113 Lymphomatoid papulosis: numerous CD30+ cells are present.
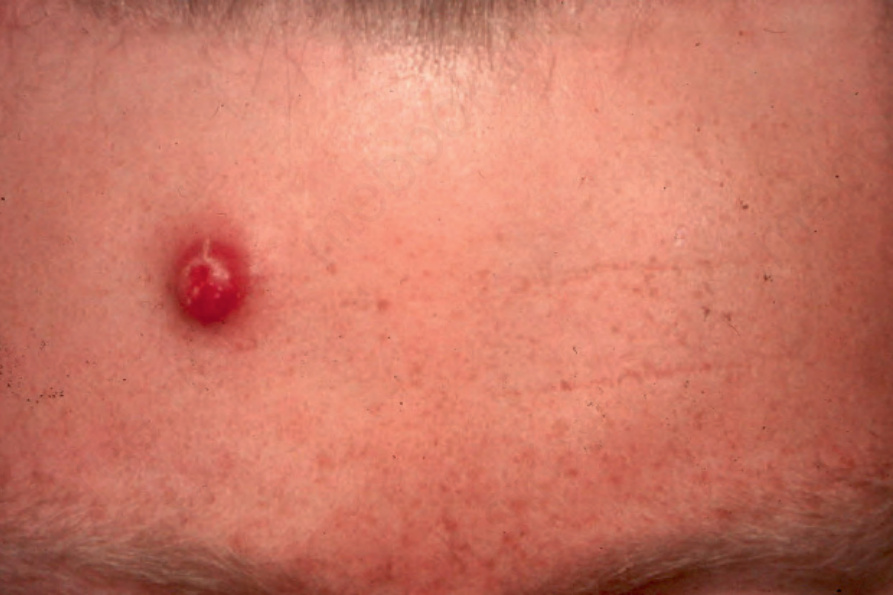
Fig. 29.114 Primary cutaneous anaplastic large cell lymphoma: erythematous, ulcerated tumor nodule on the forehead. By courtesy of the Institute of Dermatology, London, UK.
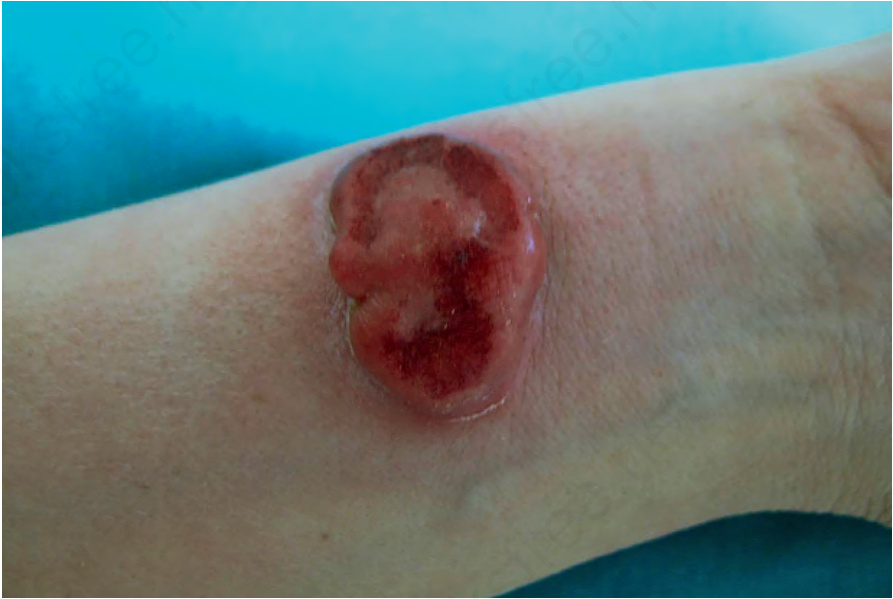
Fig. 29.115 Primary cutaneous anaplastic large cell lymphoma; large ulcerated nodule on the forearm. Courtesy of Dr Teresa Estrach Panella, Barcelona, Spain.