Sézary syndrome
Sézary syndrome
Clinical features Sézary syndrome is a rare variant of cutaneous T-cell lymphoma, accounting for <5% of cases and characterized by erythroderma, blood involvement, and a poor prognosis.1 Sézary syndrome belongs to the broader spectrum of erythrodermic cutaneous T-cell lymphomas. Also within this group are cases of erythroderma arising in patients with mycosis fungoides (erythrodermic mycosis fungoides), and cases of cutaneous T-cell lymphoma not conforming to mycosis fungoides or Sézary syndrome (erythrodermic cutaneous T-cell lymphoma, not otherwise specified). Cutaneous T-cell lymphoma with peripheral blood findings of Sézary syndrome but without erythroderma is not included, but diagnosed as ‘mycosis fungoides with leukemic involvement’.
Erythroderma is defined as a disease state in which there is diffuse erythema involving >80% of the skin surface, with or without scaling.2 It may be seen in cutaneous T-cell lymphoma as well as in a variety of other benign and malignant conditions. The latter include other lymphomas such as B-cell chronic lymphocytic leukemia, ATLL, and T-prolymphocytic leukemia.3 Benign causes include psoriasis, atopic and contact dermatitis, drug rash, and pityriasis rubra pilaris. Idiopathic erythroderma refers to cases with no identifiable etiology.2
Diagnosis is usually at least in part dependent on peripheral blood findings, and the ISCL recommends adherence to a rigorous set of criteria designed to reflect the increased blood tumor burden and poor prognosis associated with Sézary syndrome.2 These have largely been adopted in the WHO/EORTC classification of skin lymphomas1 and include;
• an absolute Sézary cell count of 1000 cells/mm3 or more,
• a CD4/CD8 ratio of 10 or higher caused by an increase in CD3+ CD4+ cells by flow cytometry,
• aberrant expression of pan-T-cell markers (CD2, CD3, CD4, CD5) by flow cytometry; deficient CD7 expression on T cells (or expanded CD4+/CD7– cells = 40%) represents a tentative criterion of Sézary syndrome,
• increased lymphocyte counts with evidence of a T-cell clone in the blood by Southern blot or PCR technique,
• a chromosomally abnormal T-cell clone. It should be noted that some peripheral blood findings may overlap with those of nonmalignant causes of erythroderma. For example, small numbers of Sézary cells may be seen in benign conditions and rarely even exceed the threshold of 1000/mm3.3 The presence of very large (diameter >14 µm) Sézary cells is a more specific but less sensitive finding.3 Some of the flow cytometric parameters are also relatively insensitive and are also occasionally seen in benign dermatoses.3–5 It may, therefore, be prudent to look for other cytometric parameters that suggest the presence of a neoplastic clone in the peripheral blood.6 These include the presence of expanded populations of CD4+CD27– T cells, CD4+CD26– T cells (with or without CD27), or an excess of T cells expressing a particular Vβ chain as part of their T-cell receptor.3,7–16 Expression of CD158 in combination with other markers may also be useful in identifying the malignant population in the peripheral blood of patients with Sézary syndrome.16–19 T-cell clones detectable by PCR or Southern blot are also rarely encountered in blood samples from patients with benign erythroderma, and can be seen as an incidental finding in the elderly.20,21 A more clinically meaningful result is obtained when an identical clone can be demonstrated in other tissues, including lymph node or skin.3,22,23
Discreet granulomas including noncaseating granulomata are sometimes a feature. The giant cells characteristically have numerous nuclei, and special stains show widespread loss of the dermal elastic tissue. A background population of small lymphocytes, plasma cells, and eosinophils may also be seen. Granulomatous vasculitis has been reported in one patient, and granulomas in the absence of overt lymphoma has been described in lymph nodes and spleen.11,20 In addition, dissemination of the neoplastic clone and associated granulomatous inflammation to lymph nodes and bronchus has been reported.11,28,30
The lymphocytes are predominantly of the helper T-cell phenotype and express CD4 and CD45RO. They may show loss or diminished expression of CD3, CD5, and/or CD7.17,20 Rare CD30-positive cells are identified. The giant cells express histiocytic markers. Many of the surrounding histiocytes can be labeled with CD1a, suggesting that they represent Langerhans cells or dermal dendritic cells.
Differential diagnosis Granulomatous slack skin shows considerable histologic overlap with granulomatous mycosis fungoides, since elastophagocytosis may be a feature of both conditions. Although the distinction is best made clinically, in granulomatous mycosis fungoides elastic tissue loss is typically focal rather than
Sézary syndrome shows a predilection for males (3 : 2) and occurs most commonly in the fifth to seventh decades.24,25 Black populations are affected twice as often as white populations.26 It is intensely pruritic and is characterized by infiltrative erythroderma (Fig. 29.96). In addition, patients often show edema of the skin and scaling, which may be particularly marked
1432 Cutaneous lymphoproliferative diseases and related disorders
Pathogenesis and histologic features The etiology of Sézary syndrome remains unknown. No consistent viral, environmental, or occupational causative factor or hereditary mutation has been found.35 Most cases show evidence of chromosomal instability with complex karyotypes and/or large numbers of chromosomal abnormalities.36–39 This genetic complexity has been confirmed in more recent genomic analyzes. These studies also demonstrate consistent genomic alterations affecting genes involved in cell cycle and epigenetic regulation as well as the T-cell receptor and JAK/STAT signaling pathways.40,41 Genomic differences between Sézary syndrome and mycosis fungoides have also been described, suggesting that these two entities may not be as closely related as previously thought.42,43
The term Sézary cell is synonymous with ‘mycosis cell’, ‘Lutzner cell’, and ‘cerebriform lymphocyte’.2 On electron microscopy, Sézary cells have characteristic hyperconvoluted (cerebriform) nuclei.44–46 They may be classified into three subtypes in peripheral blood smear according to size (Fig. 29.98):2
• small Sézary cells (Lutzner cells): 8–11 µm,
• intermediate-sized Sézary cells: 11–14 µm,
• very large Sézary cells: >14 µm. Very large Sézary cells tend only to be seen in association with lymphoma where they correlate with a worse prognosis, but it is important to remember that circulating Sézary cells of small and intermediate size may be found in a number of benign conditions including contact dermatitis, atopic dermatitis, erythrodermic psoriasis, erythrodermic eczema, actinic reticuloid, and pseudolymphomatous drug reactions.2,47–49 They may even be found in the blood of healthy elderly people.50,51
on the palms and soles (palmoplantar keratoderma) (Fig. 29.97). Hepatomegaly, alopecia, ectropion, and nail dystrophy are common manifestations, and lymphadenopathy is common.24
The outcome of the disease is variable but it is often associated with poor prognosis.24,27–33 Median survival figures of 45–48 months are often quoted, and a recent study reported a 5-year survival of 51.4%.30,32,33 Patients with visceral involvement fare particularly badly. Documented important prognostic indicators have included lymph node status, absolute Sézary cell count, fast evolution of the disease, large cell transformation and serum LDH and beta-2-microglobulin levels.30,32,33,34 The cause of death may be the tumor itself or overwhelming secondary infection. Visceral spread is similar to that of advanced mycosis fungoides.
Erythrodermic cutaneous T-cell lymphoma should be classified as stage 4 disease.
Diagnostic biopsies contain significant numbers of atypical lymphocytes with cerebriform nuclei (Sézary cells), although they are often in the minority.52,53 In 20–40% of cases these cells display epidermotropism, and the histologic features are similar to those seen in mycosis fungoides (Figs 29.99–29.101).6,32,52 In the remainder of diagnostic cases, epidermotropism is absent and the lymphoid infiltrate assumes a perivascular or superficial dermal bandlike distribution.6,32,52 Uniform small lymphocytes often predominate, but there are sufficient identifiable small, intermediate, and/or large Sézary cells to permit a diagnosis of Sézary syndrome, given the appropriate clinical setting. In a minority of nonepidermotropic cases, the dermal infiltrate consists entirely of large atypical lymphocytes and the histologic features are those of a large cell lymphoma.6,33,52 In one series, adnexal involvement was frequently seen and included follicular mucinosis in rare cases.33
Biopsies of patients with Sézary syndrome are reported to show non-specific and/or nondiagnostic features in around 40% of cases. Earlier studies using less precise diagnostic criteria may have included examples of
1433 Primary cutaneous CD30-positive T-cell lymphoproliferative disorders
this is due to accumulation of Th2-polarized neoplastic lymphocytes in the skin, resulting in reduced production of IFN-γ, leading to decreased production of IFN-γ inducible protein-10 and ICAM-1 by keratinocytes.3 Topical treatment prior to biopsy and inadequate sampling may also contribute to the frequent lack of diagnostic features. However, even when both these criteria are satisfied, the diagnosis may remain elusive.
Lymph nodes usually show features of dermatopathic lymphadenopathy but may show partial or complete effacement of the architecture by lymphoma. In contrast to mycosis fungoides, large transformed cells are not usually conspicuous.54,55
By immunohistochemistry, Sézary cells are typically CD2+, CD3+, CD4+, CD5+, CD8–, TCRαβ+, CLA+, and CD45RO+ T cells.2,33,52 CD7 is commonly diminished or absent, and CD2, CD3, and CD5 are sometimes lost.2,32 CD4–/CD8+ variants have been described, and occasionally a CD4+/CD8+ phenotype is encountered. PD1 is often positive. CD158 can be demonstrated on neoplastic cells if frozen tissue is available.56
Monoclonality may be demonstrated in skin biopsies of patients with Sézary syndrome. Such finding is best regarded as significant, particularly when an identical clone is demonstrated in peripheral blood.3,22,23
Primary cutaneous CD30-positive T-cell lymphoproliferative disorders
Primary cutaneous CD30-positive T-cell lymphoproliferative disorders are collectively the second most common group of cutaneous T-cell lymphomas, accounting for approximately 30% of cases.1,2 This category comprises a spectrum of disease with overlapping histologic and immunophenotypic characteristics, and encompasses lymphomatoid papulosis at one end and primary cutaneous anaplastic lymphoma at the other.3 Clinical appearances and disease course are critical for determining the diagnosis. ‘Borderline’ is the term applied to cases in which the distinction between lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma cannot be readily made, usually because of a discrepancy between the clinical and pathological features.2,3 Cases of CD30-positive transformed mycosis fungoides are excluded, as are other systemic CD30-positive large T- and B-cell lymphomas involving the skin.
benign erythroderma, leading to an overestimate in the proportion of such cases. However, a significant proportion of cases lack diagnostic features even in more recent studies in which Sézary syndrome was more stringently defined.4–6,32,52,53
In some studies, >50% of biopsies show features that are not diagnostic of lymphoma. These include cases with a predominantly dermal infiltrate with few, if any, recognizable Sézary cells, as well as cases showing features consistent with chronic dermatitis.19–21
Occasional nondiscriminatory additional features in diagnostic and nondiagnostic biopsies include acanthosis, parakeratosis, spongiosis, basal layer damage, and dermal fibrosis. A mixture of inflammatory cells including histiocytes, eosinophils, neutrophils, and plasma cells is sometimes seen.6,52,53
The difficulty in arriving at a histologic diagnosis has in part been attributed to a loss of epidermotropism in Sézary cells. It is speculated that
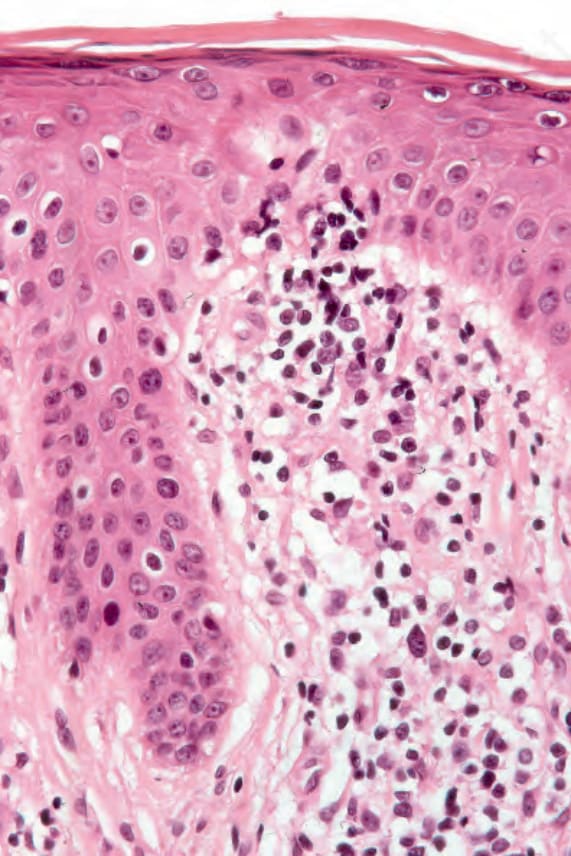
Fig. 29.100 Sézary syndrome: occasional atypical lymphocytes are present within both the epidermis and the dermis.
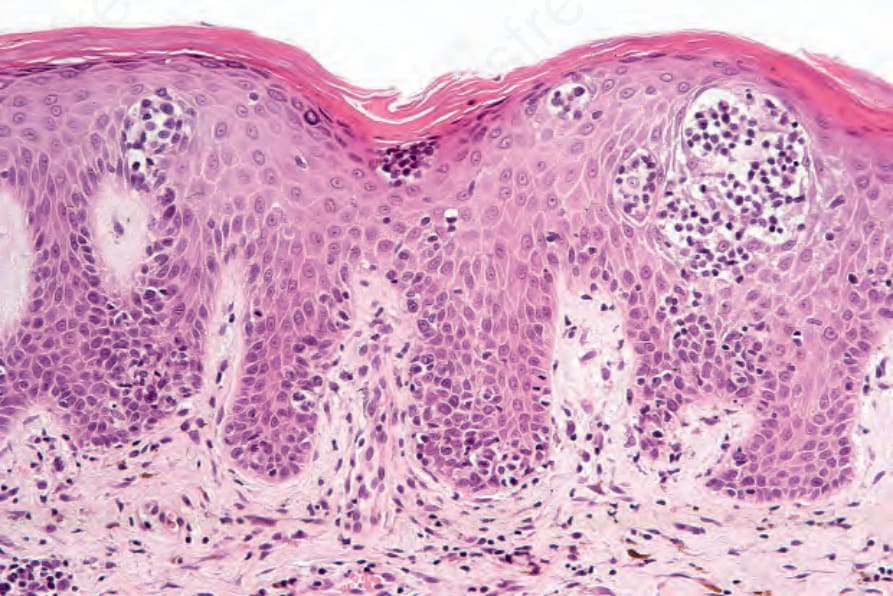
Fig. 29.101 Sézary syndrome: in this field, there are multiple Pautrier microabscesses.
Fig. 29.94 Granulomatous slack skin: there is a background population of pleomorphic lymphocytes typical of mycosis fungoides. By courtesy of P.E. LeBoit, MD, University of California, San Francisco, USA.
Fig. 29.95 Granulomatous slack skin: medium-power view showing lymphophagocytosis. By courtesy of P.E. LeBoit, MD, University of California, San Francisco, USA.
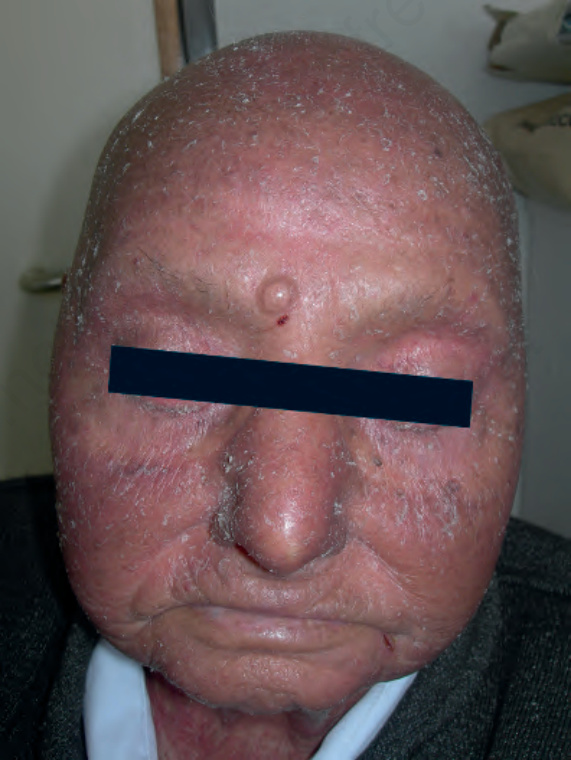
Fig. 29.96 Sézary syndrome: the facial skin is indurated and covered by a scale. Note the alopecia. By courtesy of M. Blanes, MD, Alicante, Spain.
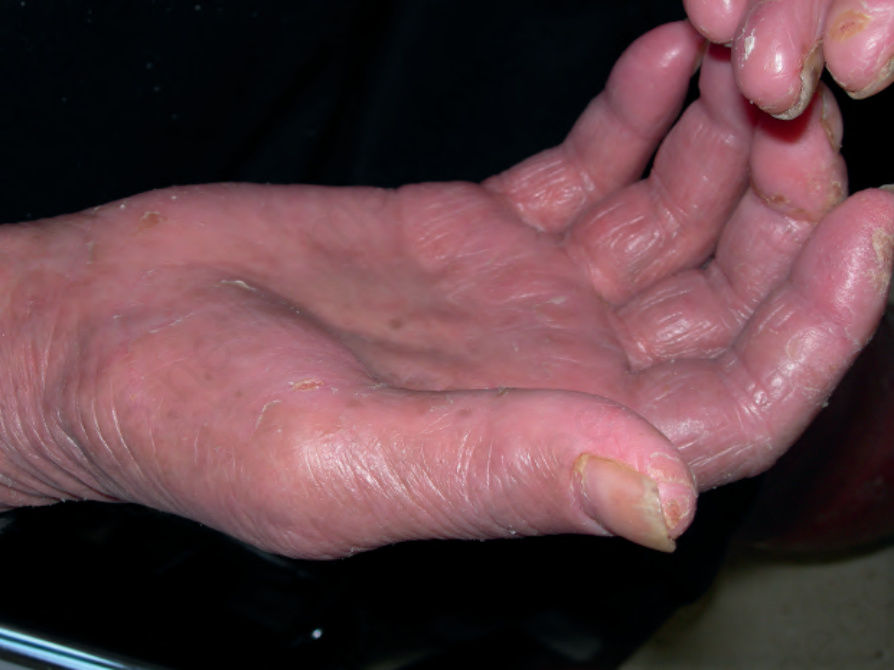
Fig. 29.97 Sézary syndrome: palmar keratoderma is often present. By courtesy of M. Blanes, MD, Alicante, Spain.
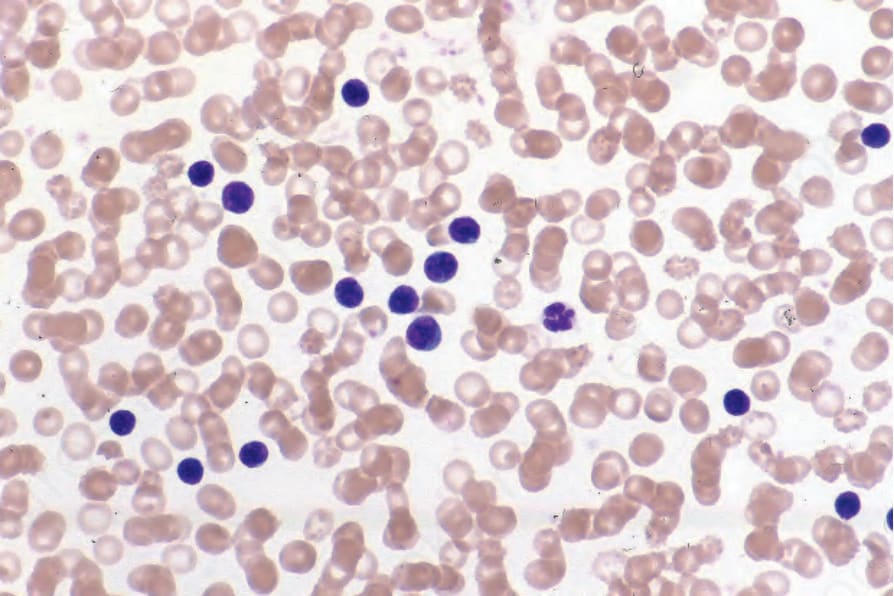
Fig. 29.98 Sézary syndrome: peripheral blood smear showing large numbers of Sézary cells. By courtesy of the Institute of Dermatology, London, UK.
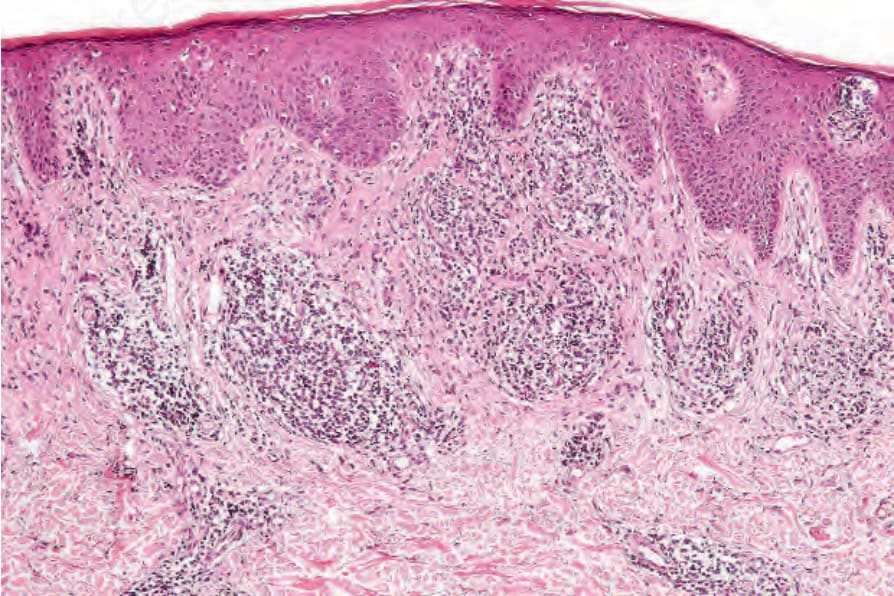
Fig. 29.99 Sézary syndrome: there is hyperkeratosis with acanthosis and a dense upper dermal lymphocytic infiltrate.