Molecular classification of melanoma — Part 4
While mutations in the receptor tyrosine kinase KIT often occur in acral and mucosal melanomas, similar to high-CSD melanomas, their incidence varies by anatomic site.34,56,58,67–69 Anorectal and vulvovaginal have the highest number of KIT mutations (approximately 35%).56,68–70 Anorectal melanomas also have frequent NF1 and SF3B1 mutations (20% each), the latter of which is often present in uveal melanomas.56 Twenty percent of acral and penile melanomas have KIT mutations58,68 while oral melanomas have approximately 10% KIT mutation prevalance.62,63 While early clinical trials of unselected melanoma patients failed to show any efficacy with KIT inhibitors,71 subsequent case reports and follow-up studies indicate that when melanoma patients with confirmed KIT mutations are selected for, dramatic responses to KIT inhibitors such as imatinib and other agents do occur.26–28,72–74 Although the majority of melanomas with genetic alterations in KIT overexpressed the protein as determined by CD117 immunohistochemistry, increased immunoreactivity does not reliably predict responding patients.39,75–77 Patients with KIT amplification without mutation do not appear to significantly benefit from the KIT inhibitor therapy.72–74
The melanoma categories in which genetic alterations of KIT are found frequently show a lentiginous growth pattern and poor circumscription
While GNAQ or GNA11 mutations are sufficient to initiate growth of these melanocytic neoplasms (Fig. 26.141), additional genomic aberrations are required for transformation into melanoma (e.g., uveal melanoma, blue nevus-like melanoma). Additional mutation or loss of the tumor suppressor BRCA1-associated protein 1 (BAP1) gene in uveal melanoma is associated with a poor prognosis and has also been found in the transformation of blue nevi to melanoma.87,88 Recurrent SF3B1 and EIF1AX mutations have also been found. They typically occur in mutually exclusive fashion from BAP1 loss and are associated with a more indolent disease course.89,90 Array CGH profiling of cellular blue nevi, atypical cellular blue nevi, and blue nevus-like melanoma shows increasing genomic instability during progression through these stages, with most nevi having no chromosomal aberrations and a
1361 Molecular classification of melanoma
features, appears to be a heterogeneous category with the majority of cases representing BRAF and NRAS mutant melanoma98 rather than melanomas that evolved from bona fide Spitz nevi or atypical Spitz tumors with HRAS mutations or various kinase fusions. The genetic features of bona fide Spitzoid melanomas, i.e., melanomas that originate from Spitz nevi or are closely related to them molecularly, are currently poorly characterized. Both homozygous loss of CDKN2A99 and TERT promoter mutation100 appear to occur in some lethal Spitzoid melanomas, though the small number of cases reported with these aberrations limits the generalizability of these findings.
Emerging concepts in metastatic melanoma The traditional concept of melanoma progression to metastasis starting from primary invasive melanoma and spreading through lymphatics to regional lymph nodes, and then from regional lymph nodes to distant sites is inadequate in explaining the observed biological behavior of metastatic melanoma. Phylogenetic analysis of separate metastatic foci in melanoma patients shows genetically distinct populations within the same patient, indicating that parallel seeding of metastatic sites occurs from the primary tumor followed by subsequent separate genetic evolution at the sites of metastasis.101–103 Metastatic ‘reseeding’, in which cells from a distant metastatic site travel to other sites of metastasis and proliferate,104 may also occur and could explain the emergence of multifocal treatment resistance to targeted therapies in some patients.105 Some evidence supporting WNT pathway activation in the progression to metastatic melanoma has been reported,101,106 and MITF amplification has been found to be more prevalent in metastatic disease.107 However, multiple genetic analyses of primary tumors and metastases have not identified a distinct set of genetic changes that reproducibly predict progression to metastasis.108–110
minority of atypical cellular blue nevi showing one to two chromosomal aberrations.88,91 Blue nevus-like melanomas exhibit multiple chromosomal aberrations (typically ≥ 3) including recurrent deletions of chromosomes 1p, 3p, 4q, 6q, 8p, 9, 16, and 17q and recurrent gains of chromosomes 6p, 8q, 20, and 21q.88,91
The Cancer Genome Atlas The Cancer Genome Atlas (TCGA) is a National Institute of Health (NIH)-sponsored cooperative effort to comprehensively analyze various cancer types. The TCGA published results on a collaborative, multi-platform analysis of 333 melanomas with a high percentage of metastatic melanomas (80%, mostly regional lymph node metastases) representing the largest study of melanoma to date.111 This study categorized melanoma by DNA mutations, copy number alterations, gene expression (RNA), non-coding RNA, methylation, protein expression, and histologic images linked to a database with clinical annotation and follow-up. In this analysis, melanomas were classified into four types: BRAF, RAS, NF1, and triple-wild type (WT). The first three types had > 90% UV signature mutation profile with high-frequency TERT promoter mutations corresponding to the low-CSD and high-CSD melanoma classes. The triple WT melanoma mutation profile was only 30% UV signature with < 10% TERT promoter mutation and had more copy number changes and fusion driver events, corresponding to the category of acral and mucosal melanomas. RNA expression analysis identified three clusters with significantly different survival rates, with the ‘immune’ signature subgroup having the most favorable survival rates and the ‘keratin’ signature subgroup showing an adverse prognosis.111 This and additional studies demonstrating prognostic value from histologic documentation of lymphocytic infiltrates as well as expression profiling of the immune response in these tumors have been reported as well.112 A separate TCGA study of primary uveal melanoma cases (n = approximately 80%) demonstrated the near universal detection of GNAQ and GNA11 mutations as well as poor prognosis associated with chromosome 3 monosomy (loss of the tumor suppressor BAP1) on chromosome 3p.90 In addition, EIF1AX and SF3B1 mutations were associated with distinct copy number alteration and methylation patterns within the chromosome 3 disomy group that corresponded to better prognosis.
Another signaling pathway implicated in the pathogenesis in tumors that share phenotypic features with blue nevi is the protein kinase A (PKA) pathway. Loss-of-function mutations in a regulatory subunit of PKA (encoded by PRKAR1A) are found in epithelioid blue nevi associated with Carney complex and result in hyperactivation of PKA with subsequent activation of the MITF transcription factor.92 Such mutations have also been detected in a subset of tumors designated as pigmented epithelioid melanocytomas.93
Spitzoid melanoma Spitzoid melanocytic neoplasms represent a class of neoplasia with a range of malignant potential from Spitz nevi at the benign end to spitzoid melanoma at the malignant end, and atypical Spitz tumors as a biologically intermediate category. Spitz nevi are defined by distinct histopathological features and an age distribution skewed towards children. They have a distinctive mutational landscape that distinguishes them from common nevi, which include activating mutations in HRAS and kinase fusions of ROS1, NTRK1, NTRK3, ALK, BRAF, RET, or MET.94–97 Spitz nevi tend to have no chromosomal aberrations with the exception of lesions with isolated chromosome 11p gain, which is typically associated with an oncogenic HRAS mutation on the duplicated chromosomal arm.94 Some Spitz nevi can also have isolated copy number increase of distal chromosome 7q, which indicates fusions of BRAF or MET mapping in this area.96,97
Atypical Spitz tumors have similar oncogenic alterations as Spitz nevi, but have some additional chromosomal aberrations, often with loss of chromosome 9. Spitzoid melanoma, when defined by solely by histologic
Summary Genetic analyses have revealed that deregulation of a few key signaling pathways is critical in melanoma formation. Common key events include activating mutations in the MAP kinase (proliferation) and PI3 kinase (survival) pathways and inactivating mutations in the RB1 pathway (cell cycle control). Kinase activation can occur at the receptor level (KIT mutations and fusions of receptor tyrosine kinases such as MET, RET, ALK, ROS1,
1362 Melanoma
NTRK1, NTRK3) often activating both the MAP kinase and PI3 kinase pathways or further downstream (NRAS, PTEN, BRAF, PI3KCA) activation of a pathway. The mutation patterns often correlate with the anatomic site of the primary tumor and clinical and histologic features. These driver mutations initiate proliferation in early stages of melanocytic neoplasia and are followed by loss of cell cycle checkpoint inhibition (e.g., p16, Rb), dysregulation of chromatin remodeling proteins (e.g., ARID1/2), and protection from replicative senescence through TERT gene amplification or TERT promoter mutation. Mutations in GNAQ/GNA11 in uveal
melanoma, blue nevi, and related tumors have highlighted additional signaling pathways of relevance in subsets of melanocytic neoplasia. The advent of high-throughput genomic tools has initiated the construction of sets of critical genetic alterations in melanoma which will in time complete the foundation for comprehensive classification of melanoma that will continue to integrate genomic alterations with established phenotypes and is expected to significantly improve prognostication and treatment stratification.
Access ExpertConsult.com for the complete list of references
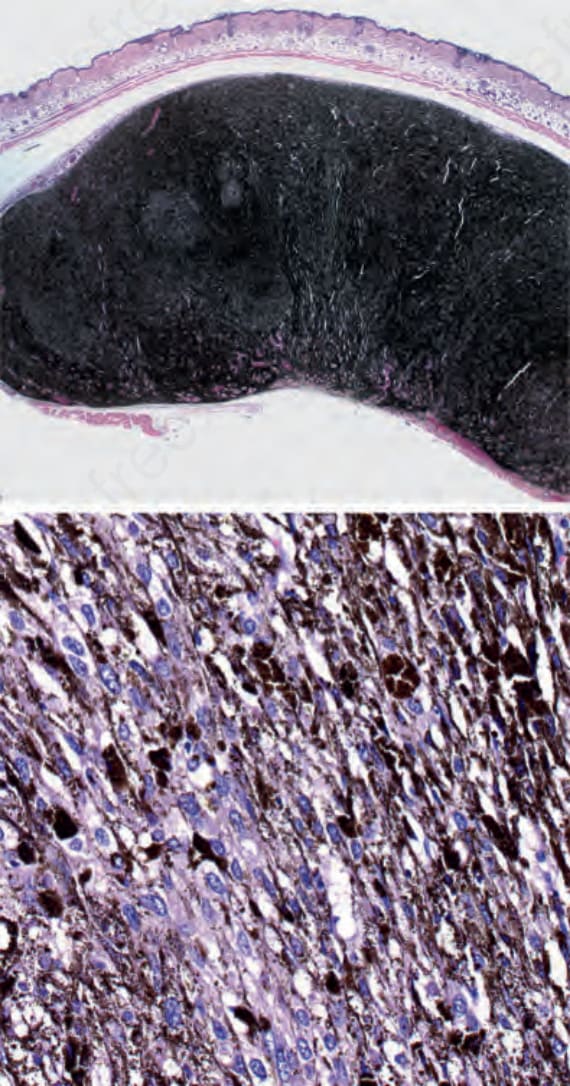
Fig. 26.141 An animal model of GNAQ mutant melanocytes: mouse melanocytes stably transduced with mutant GNAQ, but not with wild-type GNAQ, induce highly pigmented tumors of spindled and epithelioid melanocytes after 10 weeks