Molecular classification of melanoma — Part 3
Numerous other recurrent genetic alterations have been discovered in melanomas on sun-exposed skin including amplifications or mutation of MITF and activating mutations in CTNNB1, encoding β-catenin. MITF is a basic helix-loop-helix (hHLH)-leucine zipper protein that plays a role in the development of melanocytes.43 β-catenin is a critical signaling component of the WNT pathway where it acts as an adherens junction protein that can also function as a transcription factor and is mutated in a small subset of melanomas.44,45 It is also increasingly clear that telomere-related proteins are critical factors in melanoma development. Germline mutations in components of the telomere shelterin complex (e.g., POT1, ACD, TERF2IP), which is responsible for capping telomere ends and preventing recombination events, have been linked with familial melanoma.46 Mutations in the promoter of TERT, a gene encoding the protein component of telomerase, have also been described in familial melanoma and in 35% to 70% of sporadic melanomas.47,48 TERT promoter mutations are the most common mutations in melanoma and have a UV signature C > T pattern. The predisposing mutations in TERT and other telomere-related factors are thought to contribute to increasing melanoma risk by increasing telomere length and extending the replicative lifespan of neoplastic cells, thereby increasing the likelihood of acquiring the additional mutations required for transformation.49 Numerous additional genes have been implicated in melanoma, many of which are listed in Table 26.3.
Melanomas on UV-protected sites Acral and mucosal melanoma The volar surfaces of the hands and feet are minimally exposed to UV radiation, and glabrous skin has a thicker epidermis with a very thick stratum
Gene amplifications typically arise through repeated cycles of double-stranded DNA breaks and subsequent end-to-end fusions of chromatids and thus indicate a loss of control over genomic integrity. The cause of this loss of control and subsequent genetic instability is currently unclear since TP53 and other known instability-inducing mutations are uncommon in these melanomas. While it is evident that UV radiation does not play a significant role in these melanomas, it is currently unresolved whether or not there are specific carcinogens involved in the pathogenesis of acral and mucosal melanomas or whether they arise as a consequence of stochastic alterations without external influence. The marked differences in the degree of genomic instability and mutation burden between melanomas on sun-protected sites and those on sun-exposed skin indicates that the types of genetic insults are fundamentally different between these melanoma subtypes. In sun-exposed melanomas UV photoproducts and oxidative damage are the predominating drivers of tumorigenesis, whereas double-stranded DNA breaks as part of breakage-fusion-bridge cycles and/or other yet to be
2 4 6 8 10 12 14 16 18 22 Y 20
0.5 1.0 1.5 2.0
Non-CSD
0.0
-1.0
-2.0
1 3 5 7 9 11 13 15 19 21 X 17
1.0292
1359 Molecular classification of melanoma
10 mm B
A
C
D
2 4 6 8 10 12 14 16 18 22 Y 20
3
2
1
0
-1
-2
1.0292 1 3 5 7 9 11 13 15 19 21 X 17
65 0.24 sample AM108 acral
2 4 6 8 10 12 14 16 18 22 Y 20
2.0
1.0 1.5
0.5
Cyclin D1 hTERT
0.0
-1.0
-2.0
1 3 5 7 9 11 13 15 19 21 X 17
1.0292
72 0.37 sample AM124 amucosal
2 4 6 8 10 12 14 16 18 22 Y 20
2.0
1.0 1.5
0.0 0.5
-1.0
-2.0
that they reflect an early progression phase of disease.55 Once their clinical relevance has been defined, the determination of field effect may offer a more objective guidance for the size of surgical margins for removal of primary melanomas than the current empirically derived recommendations.
Although acral and mucosal melanomas share a high incidence of chromosomal aberrations, including frequent gene amplifications, the genomic regions affected by these copy number alterations differ significantly between the two.19 The most commonly amplified site in acral melanomas is the CCND1 locus encoding cyclin D1 on chromosome 11q13. Another commonly amplified site includes the TERT telomerase locus on chromosome 5p15. However, while approximately 50% of acral melanomas amplify CCND1, mucosal melanomas typically do not. Instead, a subset of mucosal melanomas amplifies cyclin dependent kinase 4 (CDK4) on chromosome 12q14, encoding the binding partner of cyclin D1.19 CDK4 is normally inhibited by p16 function, which functions as the gatekeeper of the G1/S transition point. In mucosal melanomas, the amplification of CDK4 is mutually exclusive with deletions at the p16 locus, indicating that these genomic aberrations may be functionally equivalent. Mucosal melanomas also have recurrent mutations in SF3B1, a gene commonly mutated in uveal melanoma, which is typically not found in acral melanoma.56
1.0292 1 3 5 7 9 11 13 15 19 21 X 17
discovered mechanism(s) appear critical for acral and mucosal melanoma formation.
This chromosomal instability in acral and mucosal melanoma occurs early in disease progression and has even been detected in histopathologically normal appearing melanocytes around acral melanoma in situ, so-called field cells or field effect (Fig. 26.139).55 Field cells harbor some, but not all of the gene amplifications found in the adjacent melanoma, indicating that they are precursors of the histopathologically manifest form of melanoma in situ (Fig. 26.140). These cells represent clonal expansions of melanocytes with severe genetic alterations stemming from loss of control over maintenance of genomic integrity. Such genetic changes are qualitatively different from point mutations in oncogenes such as BRAF, as they indicate loss of protective factors that preserve genomic integrity. Field cells are therefore best interpreted as an early form or precursor of melanoma in situ. These fields of neoplastic melanocytes can be extensive, present up to > 1 cm beyond the histopathologically detectable in situ portion, and the size of the field is completely independent of tumor thickness, further demonstrating
In contrast to the high number of chromosomal copy number changes and amplifications in acral and mucosal melanomas, their mutational burden is much lower than sun exposed melanomas. Many of the mutations show a pattern of mutagenesis seen with aging in which there is spontaneous deamination of 5-methylcytosine at CpG dinucleotides.31 UV signature mutations are absent for the most part. Most genetic studies of acral and mucosal melanoma focus on invasive melanoma, with very little published on the in situ stage. BRAFV600E mutation in acral melanoma in situ has been reported,57 but the incidence of BRAF mutation in acral melanoma (approximately 15–20%) is significantly lower than in low-CSD melanomas (approximately 70%) and even lower in mucosal melanoma (approximately 5%), suggesting BRAF mutation initiates a comparatively small percentage of these melanomas.34,58–63 NRAS mutation and KIT mutation or amplification are more frequently found in acral and mucosal melanoma.58,64,65 These two mutations activate the MAP kinase pathway and drive proliferation, so it is logical to assume they occur in the early proliferative phase of
1360 Melanoma
toward the uninvolved epidermis. These growth characteristics may be related to biological effects of KIT activation. KIT is essential for melanoblast migration from the neural crest into the skin and for homing into epithelial structures during development. If constitutively active KIT is introduced into human melanocytes in vitro, it induces a migratory phenotype resembling a lentiginous pattern.78 Aberrant KIT signaling in melanoma may therefore help explain the field effect described for melanomas on acral skin (Figs 26.139 and 26.140).50
Melanocytic tumors arising without associations to epithelial structures Most melanocytic neoplasms, benign melanocytic nevi as well as melanomas, originate from melanocytes situated within epithelial structures throughout the body and have mutations in genes such as BRAF, NRAS, NF1, and KIT. However, a subset of melanocytic neoplasms arises without an apparent connection to epithelial structures and does not show these mutations.38,79 One category, uveal melanoma, arises from melanocytes within the choroidal plexus, the ciliary body, and the iris of the eye and is biologically distinct from cutaneous melanoma by a very strong propensity to metastasize to the liver.80 It also differs in the presence of certain chromosomal aberrations such as frequent loss of chromosome 3, which serves as a negative prognostic indicator in uveal melanoma.81 In the skin, intradermal melanocytic proliferations, which can be congenital or acquired, present in diverse ways ranging from discrete blue papules (blue nevi) to large blue-gray patches affecting the conjunctiva and periorbital skin (nevus of Ota), shoulders (nevus of Ito), and the lower back (Mongolian spot).82 A potential connection between intradermal melanocytic neoplasms and uveal melanoma is suggested by the fact that nevus of Ota represents a risk factor for uveal melanoma in Caucasians. In addition, blue nevi can show cytological similarities to uveal melanoma.80 Interestingly, while uveal melanomas strongly express KIT on immunohistochemistry, they lack mutations in the KIT gene.79
A forward genetic screen in mice identified germline mutations in the heterotrimeric G-protein subunits GNAQ and GNA11 that cause skin hyperpigmentation by inducing a subtle intradermal proliferation of melanocytes.83 GNAQ and GNA11 are members of the q class of G-protein alpha subunits and are involved in mediating signals between G-protein coupled receptors (GPCRs) and downstream effectors.84 Somatic mutations in GNAQ are found in 83% of blue nevi, 50% of blue nevus-like melanoma (‘malignant blue nevi’), and 46% of uveal melanomas.85 The mutations in human tumors differed from the genetic variants discovered in the mouse screen. In human melanocytic tumors, all mutations occurred exclusively at codon 209, which is homologous to codon 61 of NRAS family members and leads to constitutive activation. GNAQ activates protein kinase C (PKC) family members PKCδ and ε via the release of diacylglycerol (DAG) by phospholipase Cβ.86 In vitro studies show that the GNAQQ209L mutation transforms melanocytes with efficiencies comparable to NRAS mutations and leads to activation of the MAP kinase pathway. This MAP kinase activation occurs specifically through recruitment of RasGRP3 to the cell membrane by DAG and its phosphorylation and activation by PKC δ and ɛ. RasGRP3 then activates RAS to initiate MAP kinase pathway signaling.86 When introduced into murine melanocytes, mutant GNAQ induces heavily pigmented tumors that morphologically resemble the spectrum of human blue nevi and pigmented epithelioid melanocytomas, confirming its role in driving these tumor types.85
melanoma in situ on acral and mucosal sites. Future studies are necessary to confirm this supposition. TERT promoter mutations are less common in acral melanoma (approximately 10%),58 but TERT gene amplification is frequently seen and has been documented in acral melanoma in situ.66
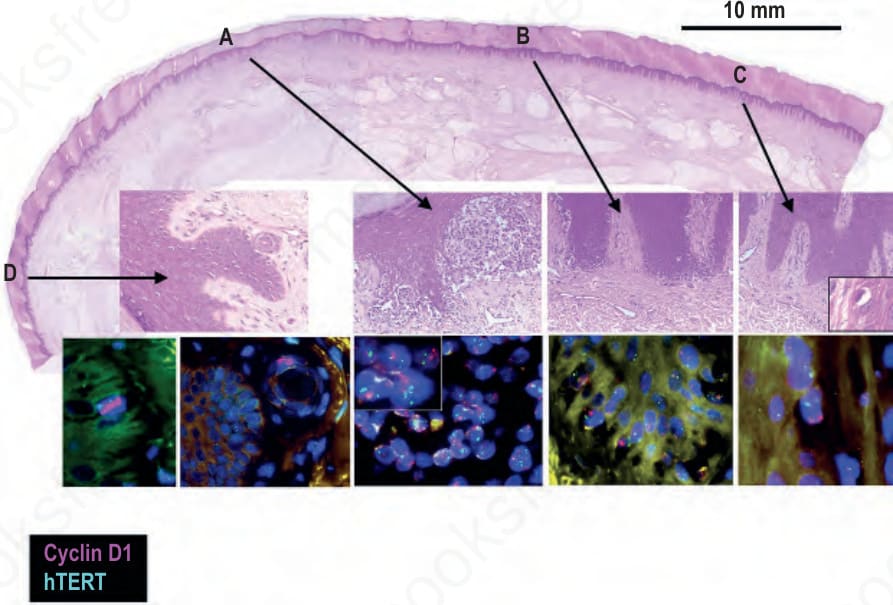
Fig. 26.139 Field cells in acral melanoma: the top panel shows an acral melanoma with close-ups of four particular areas underneath. Area A shows superficially invasive melanoma that by FISH shows amplification of CCND1 (cyclin D1) and TERT telomerase reverse transcriptase (clusters of red and green signals in blue stained nuclei, respectively). Area B shows melanoma in situ with amplification of CCND1 and normal copy number of hTERT. Areas C and D show histopathologically uninvolved epidermis which by FISH harbors single basal melanocytes with amplification of CCND1 (clusters of red signals within blue stained nuclei). In the second FISH panel for area D a melanocyte with CCND1 amplification is situated in an eccrine duct.
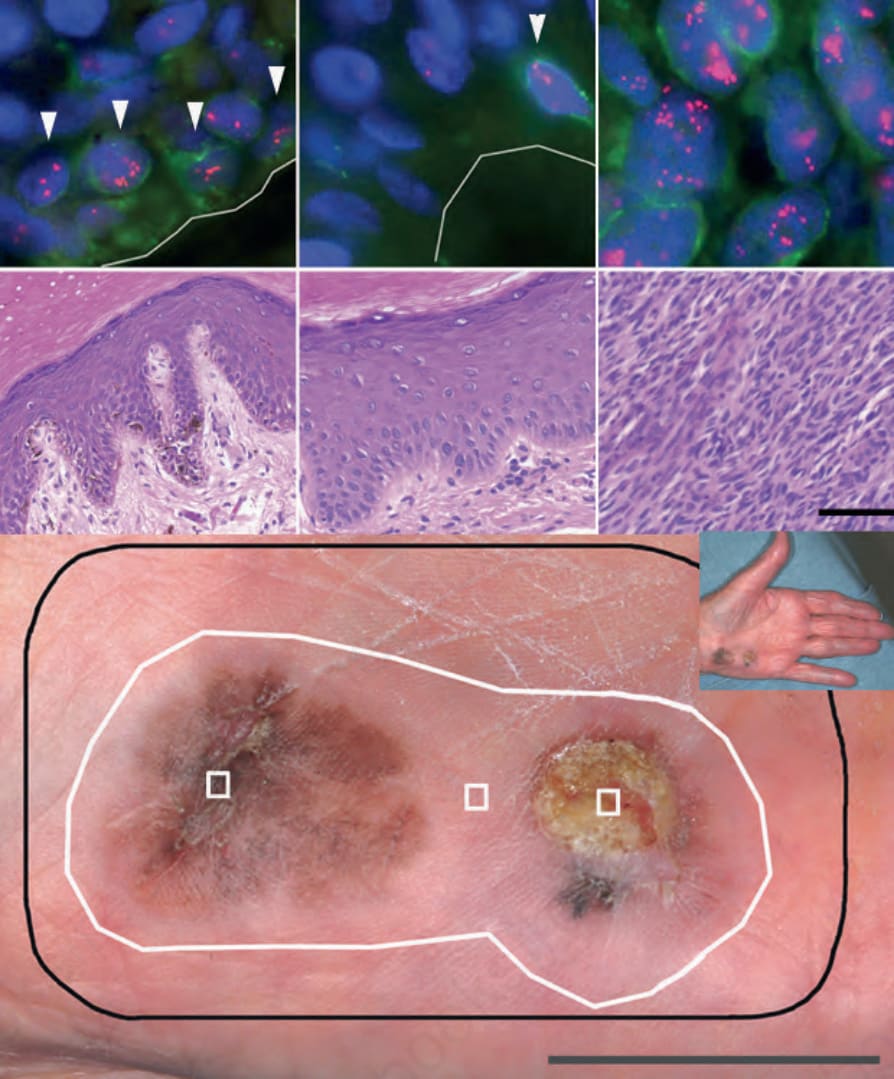
Fig. 26.140 Field cells in acral melanoma: bottom panel: clinical picture with (1) melanoma in situ; (2) clinically, dermoscopically, and microscopically normal skin; (3) invasive melanoma of 3.0-mm thickness. Black line, surgical margin; white line, field cell margin identified by amplification of 11q13. Middle and upper panels, respectively: H&E and FISH for areas 1–3. FISH images represent one focal plane, so not all signals are visible. A green immunofluorescent labeled Melan-A antibody was used to aid in identifying basal melanocytes. Cells with amplification of 11q13 are highlighted by arrowheads for areas 1 and 2 (upper panels).
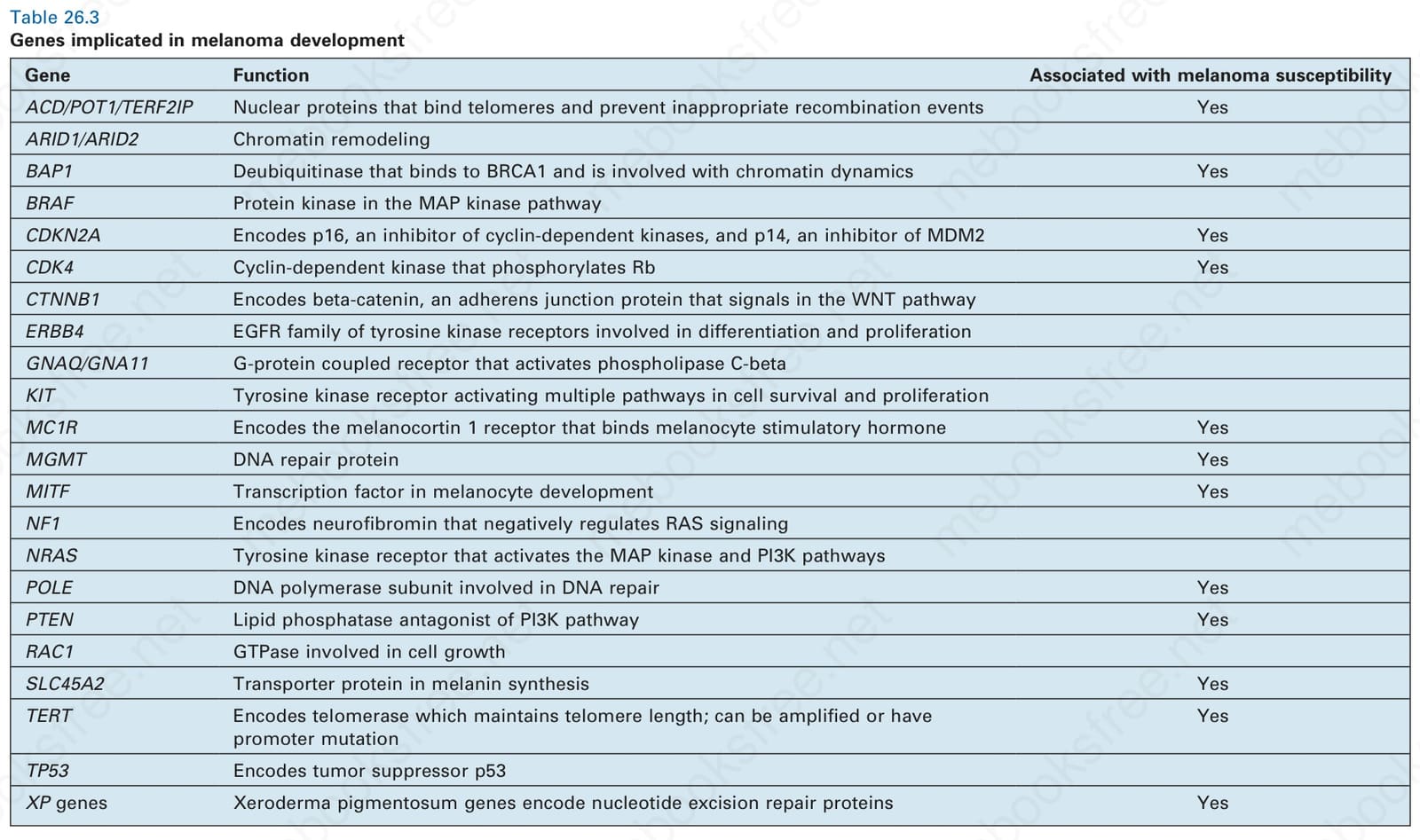
Table 26.3 Genes implicated in melanoma development