Malignant transformation of melanocytes
Malignant transformation of melanocytes
The progression of melanocyte to melanocytic nevus to dysplastic nevus to melanoma in situ and finally to invasive melanoma has served as a logical model for melanoma genesis. While this step-by-step progression occurs in some melanomas, a subset of melanomas arises without an associated nevus or in situ component (e.g., primary dermal melanoma, blue nevus-like melanoma). The number of genetic alterations necessary for melanoma formation varies depending on an individual’s inherent genetic susceptibility and the sequence and combination of genetic alterations that develop. In vitro studies using defined genetic elements indicate that as little as three different mutations are sufficient to induce melanoma-like lesions in human skin grafted on mice.12 As opposed to experimental settings, genetic alterations in real cancers typically arise sequentially. Mutations in genes driving proliferation such as BRAF, NRAS, and GNAQ/11 drive clonal expansion of melanocytes. If such initiating mutations occur in cells with intact senescence pathways, dysregulated growth triggers protective tumor suppressor checkpoints to limit proliferation and a melanocytic nevus is formed. When those proliferation-inducing mutations occur in cells harboring genetic defects that have disabled such checkpoint mechanisms, stages in the stepwise progression model can be bypassed. This is illustrated in patients with germline CDKN2A mutations who have an inherent defect in one tumor suppressor pathway. As opposed to most individuals without this mutation, these individuals develop large atypical nevi, seemingly bypassing the first benign nevus stage in which only a single pathogenic mutation (e.g., BRAFV600E) is present. It could therefore be expected that if a cell already harboring inactivated tumor suppressor pathway(s) subsequently acquired a proliferation-inducing mutation, ‘de novo’ melanoma could ensue, bypassing any phenotypically visible precursor stage.35 The combination of genetic aberrations dictates the stage of malignant transformation, with some combinations having more transforming consequences than others. For example, the combination of BRAF and BAP1 mutations appears to have low malignant potential,11 whereas combining GNAQ/11 with BAP1 mutations generates tumors with high metastatic potential.36,37
DNA damage
Apoptosis
Checkpoints
Damaged cell Arrest
Normal cell
A
Repair Normal cell
Abnormal cell Progressive genetic alterations
Malignant cell
Selection
B
Malignant
Losses Gains
n = 133
Benign
n = 54
1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19202122
0.2 0.4 0.6 0.8
Proportional aberrant
0 -0.2 -0.4 -0.6 -0.8
-1
1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19202122
0.2 0.4 0.6 0.8
Proportional aberrant
0 -0.2 -0.4 -0.6 -0.8
-1

Fig. 25.261 Barriers that restrict expansion of genetically altered cells (A) and their failure in cancer (B): (A) If checkpoints of DNA damage are intact, any cells that acquire DNA damage are transiently arrested to give time to repair the defect. If the defect cannot be repaired, cells are permanently arrested or routed to a death pathway. (B) If these checkpoints fail, cells with acquired genetic alterations can clonally expand and acquire additional genetic alterations. Over time, variants with mutations that further promote growth and survival will be selected.
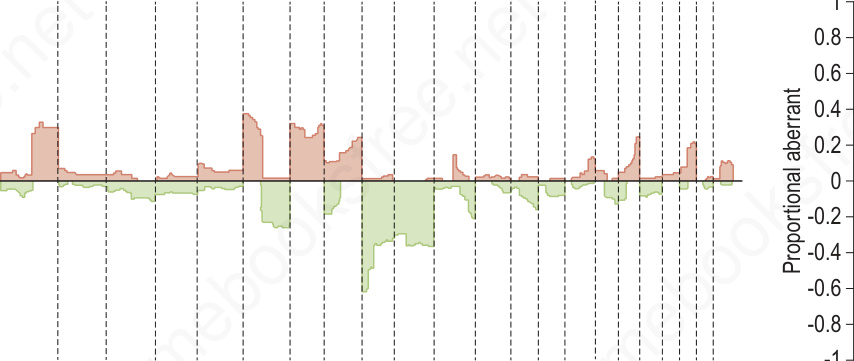
Fig. 25.262 Comparative genomic hybridization of melanoma (malignant) and nevi (benign): on the left, melanomas (n = 133) are associated with multiple copy number gains and losses that cluster. In contrast, the nevi (n = 54) on the right show minimal changes with the exception of the 11p copy number increase or amplification in Spitz nevi which includes the HRAS gene. These differences can be exploited using multiplexed FISH assays to support the diagnosis of melanoma or nevus in challenging cases.