Hypomelanosis of Ito
Hypomelanosis of Ito
Clinical features Although described here as a single entity, it is now clear that this represents pigmentary mosaicism in the setting of different diseases (see below). Hypomelanosis of Ito (also known as incontinentia pigmenti achromians) presents at birth or less commonly during infancy (about 25% of cases), with well-defined areas of macular linear or whorled hypopigmentation involving the trunk and limbs and usually (but not always or exclusively) following the lines of Blaschko (Figs 20.20 and 20.21).1,2 The hypopigmentation can be bilateral or unilateral. The areas of hypopigmentation may progress during infancy and undergo some pigmentation later in life. There is slight predilection for females, and a family history of the disease is elicited in some patients.3 The disease also occurs exceptionally in twins.4 A case associated with whorled hyperpigmentation has been described.5
Neurological and/or musculoskeletal anomalies are often associated with the disease. These include mental retardation, autism, epilepsy, seizures, macrocephaly, microcephaly, hypotonia, pes valgus, genu valgus, cerebellar hypoplasia, an intracranial arteriovenous malformation, distal spinal muscular atrophy, and hemi-overgrowth.6–8 Other more uncommon manifestations include deafness, syndactyly, clinodactyly, skeletal abnormalities, asymmetry of the facies, body or extremities, cleft palate, vesicoureteral reflux, segmental dilation of the colon, gynecomastia, cryptorchidism, inguinal hernia, short stature, oral abnormalities, congenital cardiopathies, ileal atresia, precocious puberty, and glomerulocystic disease of the kidney.6–14 Neoplasms, including neuroblastoma and coroid plexus papilloma, have also rarely been documented.15,16 An association with Moyamoya disease has been described.17
Pathogenesis and histologic features Various chromosomal abnormalities have been detected in lymphocytes and/ or skin fibroblasts including X;17 translocation and trisomy 18, trisomy 7, trisomy 2, and trisomy 13 mosaicisms.18–22
In view of the wide variety of anomalies associated with the cutaneous hypopigmentation and the variable chromosomal abnormalities, it is now believed that hypomelanosis of Ito does not represent a single disease entity but is a cutaneous sign of a group of heterogeneous disorders resulting from genetic mosaicism or chimerism.23–25
1001 Postinflammatory hypopigmentation
Histologically, there is reduction in the amount of melanin in the basal cell layer of the epidermis. The decrease in pigment occurs both in keratinocytes and melanocytes and the change is easy to evaluate in biopsies stained by Masson-Fontana. Decrease in the number of melanocytes does not seem to be a feature but has been reported occasionally. Ultrastructurally, melanocytes may show vacuolization and decrease in size and in the number of dendrites.26,27 Increased numbers of epidermal Langerhans cells have been documented.28
Differential diagnosis Distinction from incontinentia pigmenti is based on the absence of melanophages in the papillary dermis in hypomelanosis of Ito.
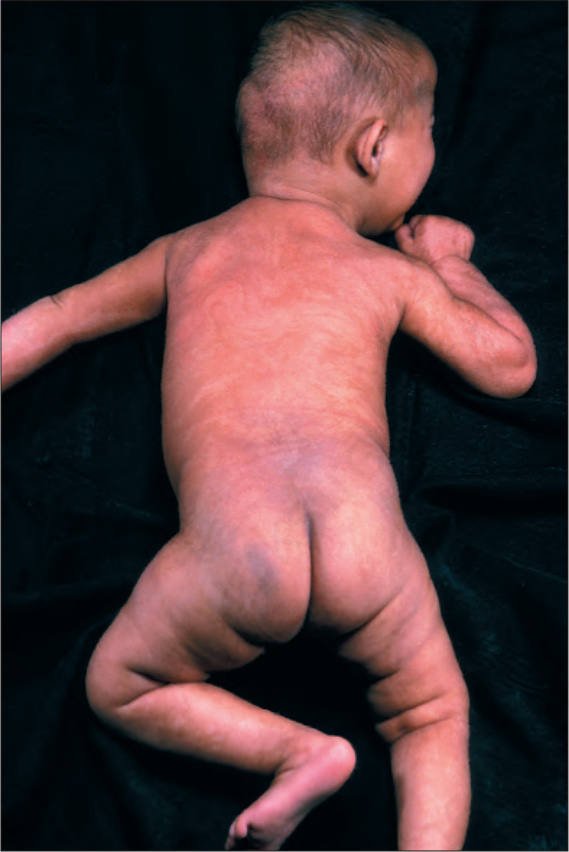
Fig. 20.20 Hypomelanosis of Ito: whorled hypopigmentation on trunk and limbs. By courtesy of the Institute of Dermatology, London, UK.