Oculocutaneous albinism
Oculocutaneous albinism
The term ‘albinism’ is used to refer to a heterogeneous group of autosomal recessive conditions resulting from diverse genetic abnormalities that cause absent or reduced production of melanin pigment but no reduction in the total number of melanocytes (Fig. 20.11).1,2 The genetic abnormality may result in eye involvement only (ocular albinism) or may combine involvement of the eyes, hair, and skin (oculocutaneous albinism). Here, only the different types of oculocutaneous albinism will be discussed.
At least 15 different gene mutations leading to different types of albinism have been identified.3 The prevalence of oculocutaneous albinism is around 1 : 17 000.4 The clinical manifestations of oculocutaneous albinism depend on the type of biochemical abnormality. In all types of albinism, the lack of melanin in the developing eye results in hypoplasia of the fovea and abnormal routing of the optic nerves. As a result, all patients have variable strabismus, nystagmus, and reduced visual acuity.4 Affected patients often have an increased risk of skin cancer (Figs 20.12 and 20.13). Based on clinical features alone, it is not always possible to separate the different types of oculocutaneous albinism.
Pathogenesis and histologic features The exact pathogenesis of idiopathic guttate hypomelanosis remains unknown. It is likely that the cause of the disease is multifactorial. Factors that have been involved in its causation include inheritance, ultraviolet (UV) light, and loss of melanocytes due to aging in addition to autoimmunity.4–7,10–12 An association with HLA-DQ3 was found in a group of renal transplant patients with the disease while a similar group of renal transplant patients without the disease had HLA-DR8, suggesting that the latter may have a protective effect.13
The main histologic feature consists of variable loss of melanin granules in epidermal keratinocytes. This is sometimes associated with epidermal atrophy and flattening of the rete ridges. Orthokeratotic hyperkeratosis in a basket-weave pattern has also been documented.7 A Masson-Fontana stain is useful to highlight the loss of epidermal melanin, and a dopa oxidase reaction reveals a patchy reduction in the number of melanocytes.2 However, complete loss of melanocytes is not a feature. Hypopigmented keratosis
The most common types of albinism are oculocutaneous albinism types IA and II. Most types are inherited in an autosomal recessive manner and affect all races.5,6 Prenatal diagnosis of oculocutaneous albinism is possible.7,8 Rare variants of albinism are associated with systemic manifestations and include the Hermansky-Pudlak and the Chédiak-Higashi syndromes (see below). The different types of oculocutaneous albinism and the histopathology of all variants are discussed briefly.
995 Oculocutaneous albinism
Pathogenesis In all variants of albinism melanocytes and melanosomes are normal. The reduction or absence of melanin results from a biochemical abnormality in the synthesis of the pigment. The gene for tyrosinase has been cloned to chromosome 11q14.3. Numerous different mutations in the gene lead to absence of tyrosinase activity and the manifestations of the disease.4,15–18
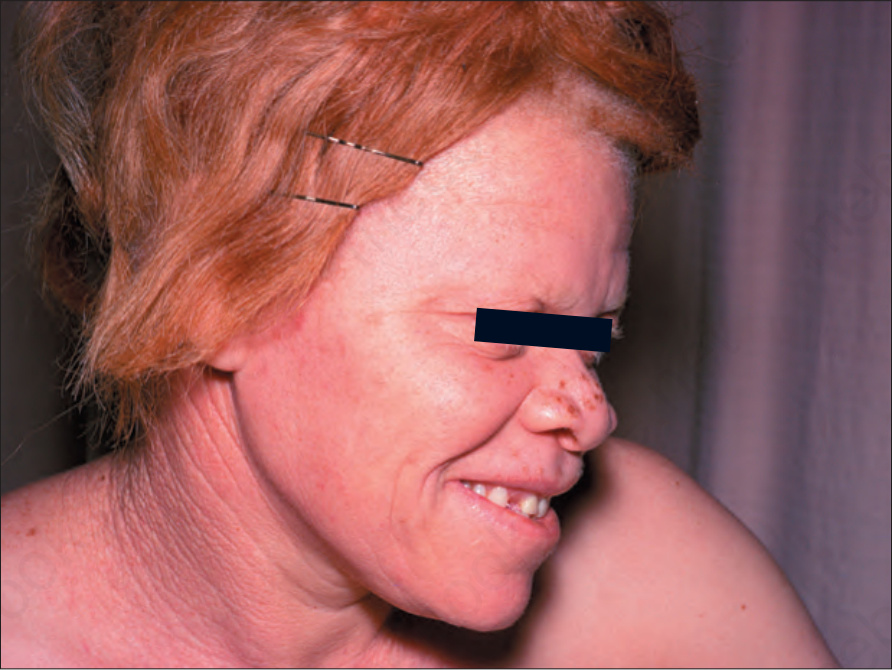
Fig. 20.11 Oculocutaneous albinism: the hair is red and there is complete loss of skin pigmentation with some freckling and actinic damage. By courtesy of the Institute of Dermatology, London, UK.
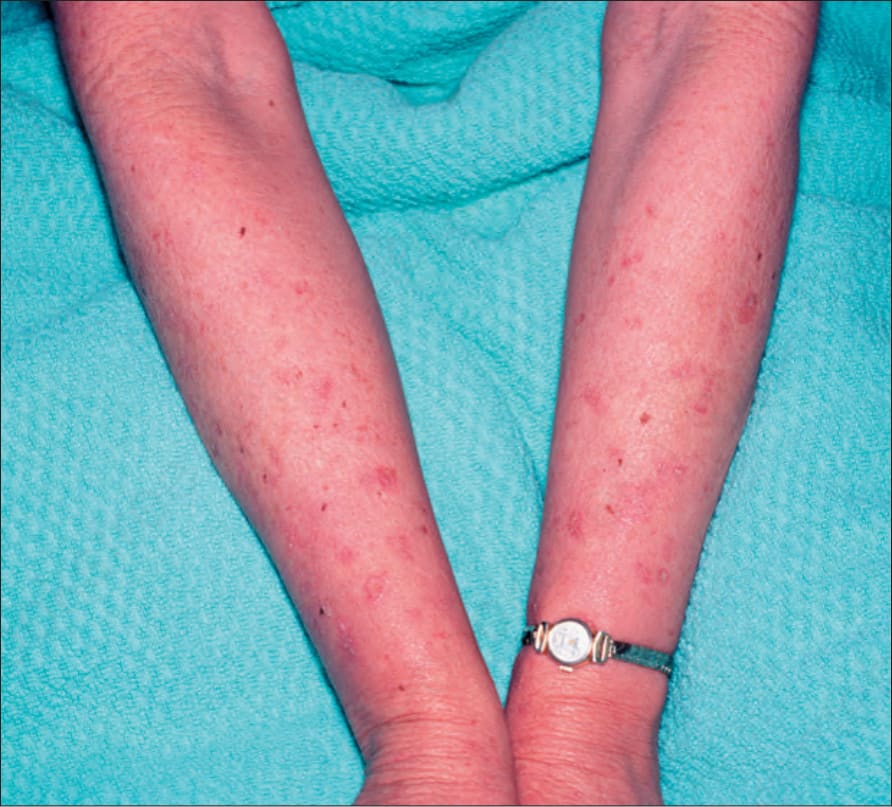
Fig. 20.12 Oculocutaneous albinism: numerous actinic keratoses on sun-exposed skin. By courtesy of the Institute of Dermatology, London, UK.
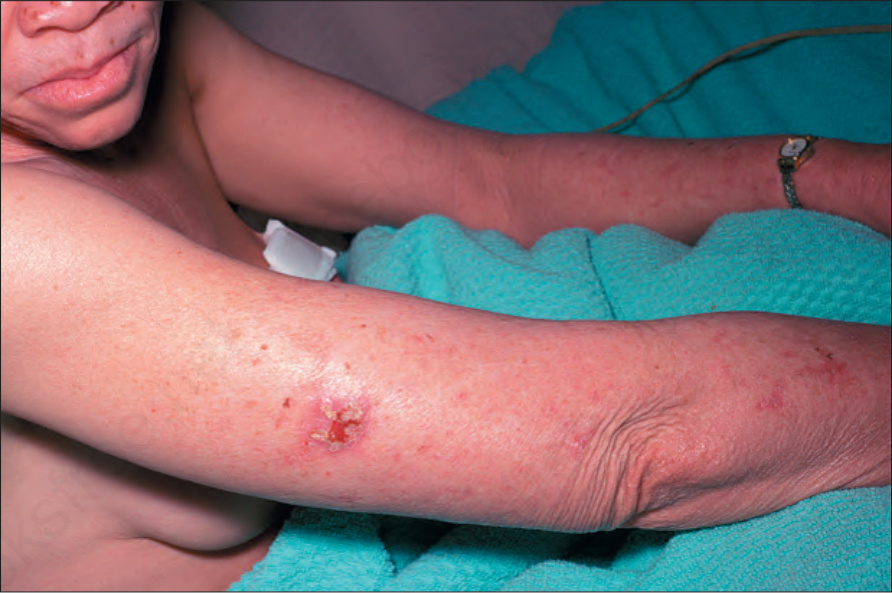
Fig. 20.13 Oculocutaneous albinism: an early squamous cell carcinoma on sun-exposed skin. By courtesy of the Institute of Dermatology, London, UK.