Vogt-Koyanagi-Harada disease
Vogt-Koyanagi-Harada disease
Clinical features Vogt-Koyanagi-Harada (VKH) (uveoencephalitis) disease is characterized by bilateral uveitis, poliosis, vitiligo, alopecia, dysacousia, and aseptic meningitis.1–5 It is more common in females (2 : 1), particularly from Latin America, Asia, and Africa, and in Native Americans. It shows a predilection for adults, occurring only rarely in children. The initial phase of the disease usually starts with neurological symptoms and this is followed by ocular manifestations. In the acute phase, exudative retinal detachment is quite specific.6 As the disease progresses, patients develop cutaneous manifestations with focal or diffuse alopecia areata, vitiligo (mainly head and trunk), and poliosis (eyebrows, lashes, and hair) in the chronic stage of the disease. In the latter phase, sunset glow fundus is often found.6 It has been suggested that VKH disease is part of the spectrum of vitiligo with manifestations in other melanocyte-containing organs.1 Not every patient displays all the manifestations of the syndrome. Ocular complications consist mainly of glaucoma and cataracts.2,3 Rarely, the patches of depigmentation have an inflammatory edge.7,8 The disease is exceptionally preceded by erythroderma.9
Histologic features Microscopic examination of involved skin in vitiligo shows complete absence of melanocytes in association with total loss of epidermal pigmentation (Fig. 20.8). Biopsies from the periphery of lesions with a clinically inflammatory border show lymphocytes and histiocytes in the papillary dermis. Some inflammatory cells may also be found at the periphery, even in the absence of a clinically inflamed border. A prominent mononuclear inflammatory cell infiltrate is exceptional.118 Degenerative changes have been documented in keratinocytes and melanocytes, not only from the border of lesions, but also from adjacent skin.119,120 Degenerative changes in nerves and sweat glands have also been documented.121 Absence of Merkel cells in lesional
994 Disorders of pigmentation
VKH disease has been described in association with diabetes, hypothyroidism, melanoma, ulcerative colitis, Graves disease, psoriasis, brainstem encephalitis, tuberculosis, bacille Calmette-Guérin (BCG) and influenza vaccination, and seronegative rheumatoid arthritis.10–18 A possible association with combination therapy with IFN-α 2b/ribavirin and with ipilimumab and pembrolizumab for metastatic melanoma have been documented.19–21
Pathogenesis and histologic features As with vitiligo, the pathogenesis of the disease remains unknown. An autoimmune etiology in which melanocytes are targeted is favored;22 however, it is likely that the etiology is multifactorial. This is based on the occurrence of the disease in identical twins and after cutaneous injury.23,24 An infectious etiology – particularly a virus – has been suggested in the past and this is supported by the simultaneous onset of the syndrome in a group of six patients living and working together.25 An association with human leukocyte antigen (HLA)-DR53, HLA-DQ4, HLA-DR1, HLA-DR *0405 in Saudi Arabian patients, and HLA-DR4 in Brazilian patients suggests a genetic predisposition.26,27 A study has raised the possibility that the disease is triggered by synergistic hyporesponsiveness of NK cells and cytotoxic T lymphocytes resulting in failure to mount an effective immune response as a result of a viral infection in genetically susceptible individuals (e.g., HLA-DR4 carriers).28 A similar disease has been induced in rats by tyrosinase-related proteins TRP1 and TRP2, suggesting a role in the pathogenesis of the disease.29 Increased susceptibility to the disease has been suggested in Han Chinese as a result of hypermethylation of GATA3 and transforming growth factor (TGF)-ß.30 Susceptibility loci have been identified at chromosomes 1p31.2 and 10q21.3.31
The histopathology of the areas of cutaneous depigmentation is identical to that seen in vitiligo. In a single case report of a patient presenting with inflammatory vitiligo, filamentous masses and amyloid were described.8 The histologic findings in areas of alopecia are identical to those seen in alopecia areata.32 However, more prominent release of melanin has been described, suggesting that the prime targets are the melanocytes within the hair follicle rather than the keratinocytes, as in classic alopecia areata.32
has been proposed as a variant of guttate hypomelanosis with prominent hyperkeratosis.14
Electron microscopy studies confirm the reduction in the number of melanocytes. Changes described in melanocytes include cytoplasmic vacuolization and decrease or loss of mature melanosomes.5,6,11,15 Although the number of Langerhans cells is normal, a reduction in the number of Birbeck granules has been documented.10
Differential diagnosis The clinical differential diagnosis is very wide and includes lichen sclerosus, hypopigmented mycosis fungoides, atrophie blanche, vitiligo, pityriasis versicolor, leprosy, and the hypopigmented lesions seen in tuberous sclerosis. Most of these entities are easy to exclude histologically except for vitiligo. In the latter, there is complete absence of melanocytes and this can be confirmed with a melanocyte-specific immunostain such as Melan-A, since S-100 also stains Langerhans cells.
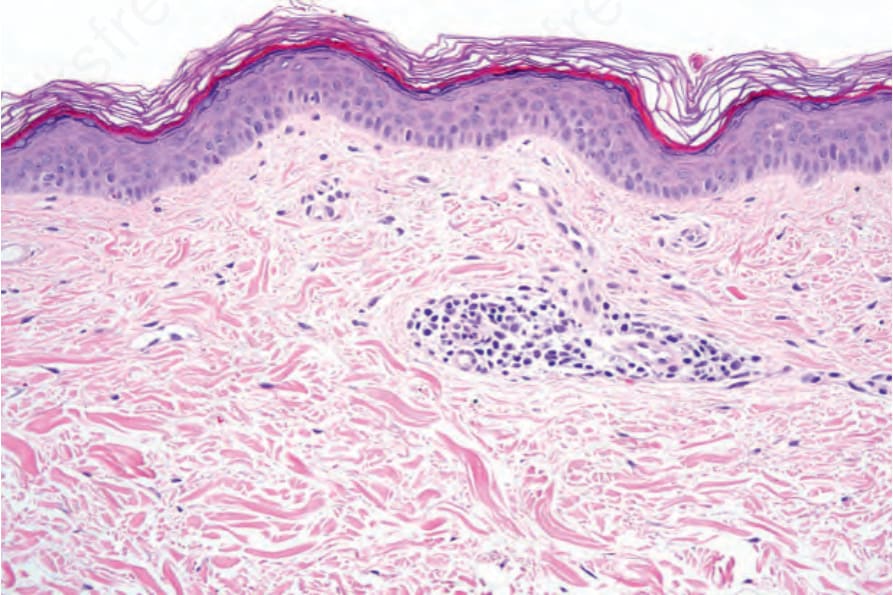
Fig. 20.8 Vitiligo: skin biopsy from lesional skin stained with hematoxylin and eosin. There is complete absence of melanocytes and melanin pigment. There is a light perivascular chronic inflammatory cell infiltrate.
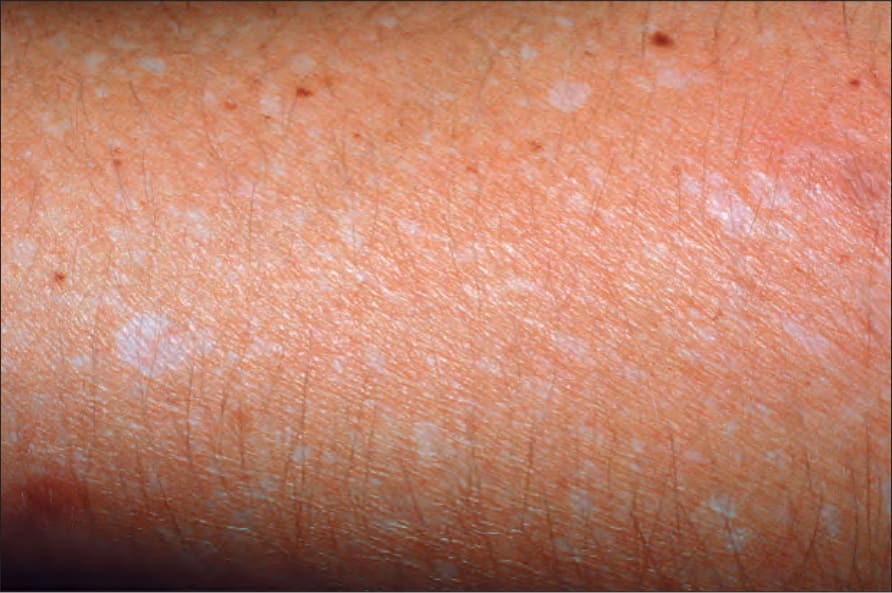
Fig. 20.10 Guttate hypomelanosis: multiple small hypopigmented macules in sun-exposed skin. By courtesy of O. Dueñas, MD, Bogotá, Colombia.