Relapsing polychondritis
Relapsing polychondritis
Clinical features Relapsing polychondritis is a rare disorder characterized by recurrent episodes of inflammation of cartilaginous tissue throughout the body and its subsequent degeneration and replacement by fibrous tissue (Fig. 17.142).1,2 The ears (93%), nose (56%), larynx, and trachea (30%) are predominantly affected.3–7 Skin manifestations are the presenting features in approximately 50% of cases.6 There is a slight male predominance and the median age
at diagnosis in one large study was 46.6 years.8 Presentation in children is exceptional.9,10 Pediatric and adult-onset relapsing polychondritis patients share similar clinical features.10 However, children have a family history of autoimmune diseases more often than adults. However, they infrequently present with associated autoimmune conditions.10 Clinical criteria for
824 Idiopathic connective tissue disorders
Recurrent articular chondritis Cochlear and vestibular damage Ocular involvement Nasal involvement Tracheal/pharyngeal involvement Nonerosive polyarthritis
MCTD, SLE, inflammatory bowel disease (both ulcerative colitis and Crohn disease), and myeloproliferative disorders.2,6,34,48–56 Rare associations of relapsing polychondritis include ankylosing spondylitis, Behçet disease, HIV, splenic abscess, chronic hepatitis C, mixed cryoglobulinemia, sarcoidosis, common variable immunodeficiency, familial Mediterranean fever, synovial chondromatosis of the temporomandibular joint, immunoglobulin G4-related disease, and amyloidosis.57–67
An increased ESR and anemia are the commonest significant laboratory manifestations. Increased urinary glycosaminoglycans have also been documented.68
diagnosis have been established. Three of these, together with biopsy confirmation of chondritis, are required for diagnosis (Table 17.15).5
Although it particularly affects Caucasians, cases have been recorded in Asians, blacks, Hispanics, and the Japanese.3 The sex incidence is equal. Most patients present in the fourth and fifth decades of life.4,11 There is no evidence of a hereditary predisposition.6 Clinical signs may be subtle and can resemble those seen in Behçet disease or inflammatory bowel disease; the diagnosis is often difficult.2,12 Familial cases are exceptional.13 Variabilities in the disease presentation have been reported in different ethnic populations. The incidence of relapsing polychondritis has been estimated to be 3.5 per million per year.14
Auricular chondritis is the commonest lesion and is frequently bilateral.3 Patients present with painful, tender, erythematous, sometimes blue-black, and swollen ears.6 Chronicity leads to distortion and flabbiness. Arthritis (seronegative) particularly affects the sternoclavicular, costochondral, and sternomanubrial joints.3 One or more joints may be affected and lesions are often migratory.6 Painful nasal chondritis may result in epistaxis, and saddle nose is an occasional complication. Nasal involvement is seen in over 50% of patients.8 Oral aphthosis was present in 11% of patients in a large series.2 In 6% of patients, oral and genital aphthae were seen.2 When the disease initially presents, inflammation of a single site may be confused with erysipelas.15
Relapsing polychondritis has a significant mortality. The 5-year survival rate is approximately 74%.34 Infection, respiratory failure, systemic vasculitis, large vessel aneurysm rupture, and renal failure are the commonest causes of death.1,34
A number of patients have been reported to have myelodysplastic syndrome associated with relapsing polychondritis.43,69–73 This is of interest since myelodysplastic syndrome is known to be associated with autoimmune disease.74,75 In a large study of 200 patients, 11% had myelodysplastic syndrome.2 Other malignancies occurring with relapsing polychondritis include splenic non-Hodgkin lymphoma, chronic lymphocytic leukemia, chronic myelomonocytic leukemia, and Kaposi sarcoma.76–79 Association with epithelial malignancies is less frequent.80
Relapsing polychondritis has been associated with the luteinizing hormone-releasing hormone (LH-RH) analog goserelin. As the disease may worsen during pregnancy and during chorionic gonadotropin therapy, it is suggested that hormones may be a precipitating factor.81
Ocular lesions include conjunctivitis, corneal ulceration, iridocyclitis, episcleritis, proptosis, cataract, chorioretinitis, scleromalacia perforans, scleritis, retinal detachment, blindness, edema of the eyelids and muscle palsies, and optic neuropathy.3,6,8,16–19 Chronic conjunctivitis due to obliterative microangiopathy has been reported in a single patient with relapsing polychondritis.20 Central nervous system complications comprise aseptic meningitis and meningoencephalitis, encephalitis lethargica, Lewy body-like dementia is a rare complication.19,21–25 Trigeminal neuralgia has also been reported.26
Pathogenesis and histologic features The precise etiology of relapsing polychondritis is poorly understood. Several studies have suggested an immunological mechanism.6 The association with autoimmune diseases in many patients lends support to this thesis. Antibodies (predominantly IgG) to type II collagen, which accounts for over 50% of the proteins in cartilage, have been detected in a proportion of patients in titers of 1 : 10 to 1 : 320.39,48,82,83 The antibodies are directed against both native and denatured protein.48 Using ELISA, one study showed that 50% of patients have antibodies against type II collagen.84 In this same study, 4% of control subjects and 15% of rheumatoid arthritis patients also had antibodies in their sera. Those patients who have the autoantibody show evidence of active disease, whereas those without it are either in remission or being treated.48 Rats immunized with type II collagen develop auricular chondritis. Cartilage from these same animals had positive immunofluorescence for IgG and C3.85 In a single patient, T-cell clones were found to be specific for the collagen II peptide 261–273.86
Respiratory lesions may affect the larynx, trachea, and major bronchi with obstructive symptoms, stenosis, collapse, pneumothorax, pneumoperitoneum, and bronchopneumonia.3,7,8,27 Exceptionally, airway involvement may be the only manifestation of the disease.28 Cardiovascular lesions include valvular incompetence, conduction defects including complete heart block, cystic medial necrosis of the aorta, aortitis, aortic valve regurgitation, vasculitis and pericarditis, and pericardial effusions.3,8,19,29–32 Involvement of the heart valves occurs in up to 10% of patients and systemic vasculitis, reminiscent of polyarteritis nodosa, has been described.33 Ear involvement includes external ear chondritis, otitis media, vertigo, and deafness.34
An association between the disease and HLA-DR4 has been reported in a study from Germany but there was no predominance of any DR4 subtype.87 Nevertheless, a recent population-based study from Japan failed to confirm the association between relapsing polychondritis and HLA-DR4, but instead confirmed association between the presence of HLA-DRB116 : 02, HLA-DQB105 : 02, and HLA-B*67 : 01 in Japanese relapsing polychondritis patients.88
Antifetal cartilage antibodies have been detected by indirect immunofluorescence studies.83 Documentation of transplacental transfer of these antibodies with neonatal involvement suggests that they are of pathogenetic significance. One group of authors suggested that matrilin-1, a cartilage matrix protein, is the target of autoreactivity.89,90 Another group found autoantibodies to matrilin-1 in 13% of patients and antibody titers correlated with symptomatology.91 Rats immunized with matrilin-1 develop nasorespiratory abnormalities (but not ear or joint changes).92 Cartilage oligometric matrix protein has also been suggested as a potential autoantigen.93
Dermatological manifestations are present in 35% to 50% and may even precede the development of relapsing polychondritis in about 12% of cases.2 Skin lesions have included leukocytoclastic vasculitis, hypocomplementemic urticarial vasculitis, cutaneous polyarteritis nodosa, erythema elevatum diutinum, livedo reticularis, alopecia, retarded nail growth, erythematous nodules, erythema annulare centrifugum, erythema multiforme-like lesions, urticarial plaques, erythema nodosum, thrombosis, pyoderma gangrenosum-like lesions, Sweet syndrome, postinflammatory hyperpigmentation, and psoriasis.2,3,6,35–45 Exceptional associations with normolipemic plane xanthomatosis and with panniculitis showing septal and lobular involvement accompanied by vasculitis have been documented.46,47
Significant disease associations that may be present in up to 30% of patients include leukocytoclastic vasculitis, systemic vasculitis (Takayasu and temporal arteritis, Wegener granulomatosis), p-ANCA associated vasculitis, Hashimoto thyroiditis, arthritis, Sjögren syndrome, dermatomyositis,
Circulating immune complexes have also been demonstrated in relapsing polychondritis, together with deposits of immunoglobulin and complement in inflamed cartilage, adding further support to a possible immune mechanism in this disease.3,40,48 Granular deposits of immunoglobulin and complement (C3) have been described at the chondrofibrous junction in two patients.93 The presence of ANCA has been reported.94 Elevated serum levels of macrophage migration inhibitory factor have also been documented.95 Increased serum levels of proinflammatory cytokines, namely
825 Relapsing polychondritis
macrophage inflammatory protein 1β, monocyte chemoattractant protein 1, and interleukin-8, have also been demonstrated in patients with relapsing polychondritis.96
There is some evidence suggesting that cell-mediated immunity may also be of importance in the pathogenesis. Patients display positive lymphoblast transformation and macrophage migration inhibition to cartilage glycosaminoglycans.1 Responses correlate with episodes of disease activity. Dysregulation of NKT cells has also been detected in relapsing polychondritis.97
Histologic examination of the skin is unremarkable. The dermis contains a mild focal lymphohistiocytic infiltrate. Examination of the fibrocartilaginous tissues, however, shows degenerative and inflammatory changes affecting the marginal chondrocytes, with loss of basophilia and poor Alcian blue staining of the cartilaginous tissue (Figs 17.143 and 17.144).
The inflammatory cell infiltrate, which includes lymphocytes, plasma cells, histiocytes, and occasional polymorphs, infiltrates the degenerate cartilage. Eventually, there is replacement by granulation and fibrous tissue.3 Atypical lymphoid infiltrates mimicking a lymphoma have rarely been described.98
Differential diagnosis Chondrodermatitis nodularis helicis differs by the presence of characteristic layering of fibrin, granulation tissue, and cartilage with degenerative changes. Clinically, chondrodermatitis helicis presents as a focal, punched-out ulcer. This differs from the diffuse involvement of the ear seen in relapsing polychondritis.
Access ExpertConsult.com for the complete list of references
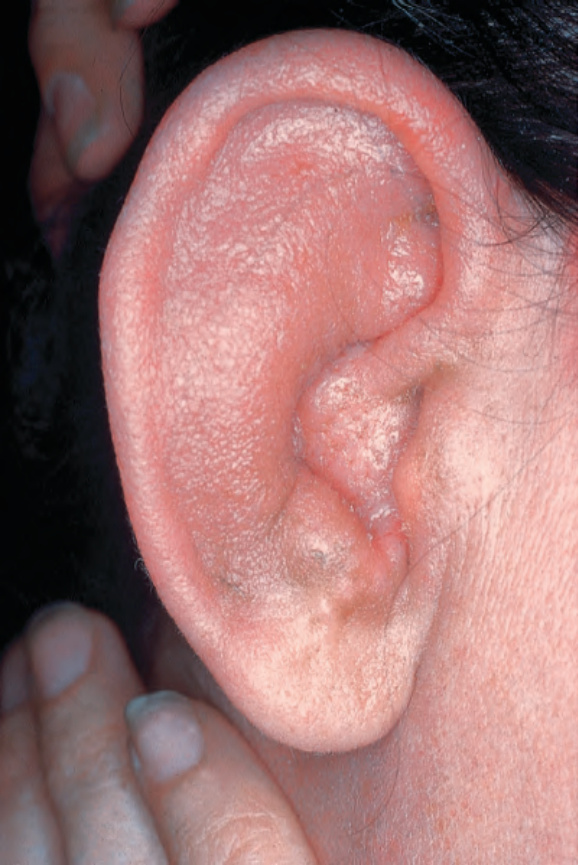
Fig. 17.142 Relapsing polychondritis: the ear shows considerable erythema and swelling. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
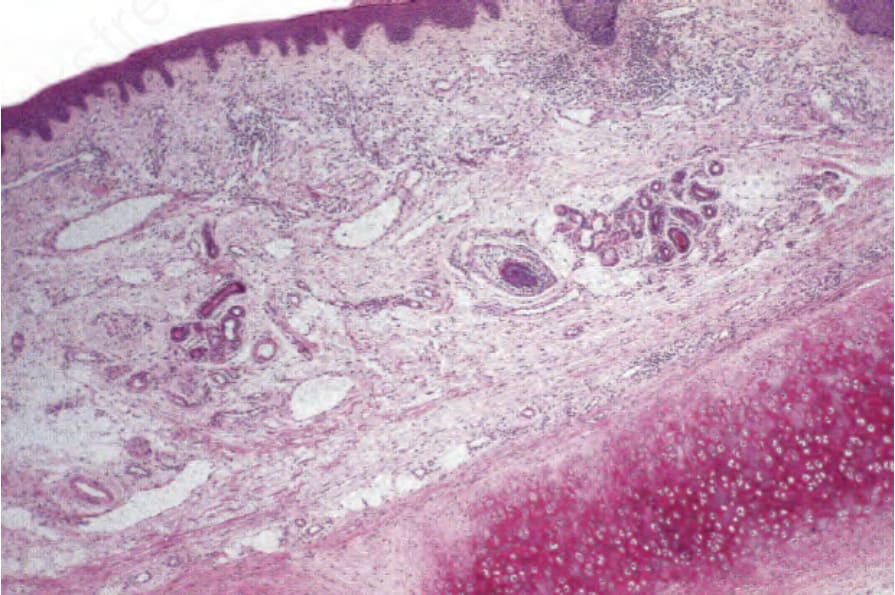
Fig. 17.143 Relapsing polychondritis: in this early lesion, the degenerate cartilage shows intense eosinophilia.
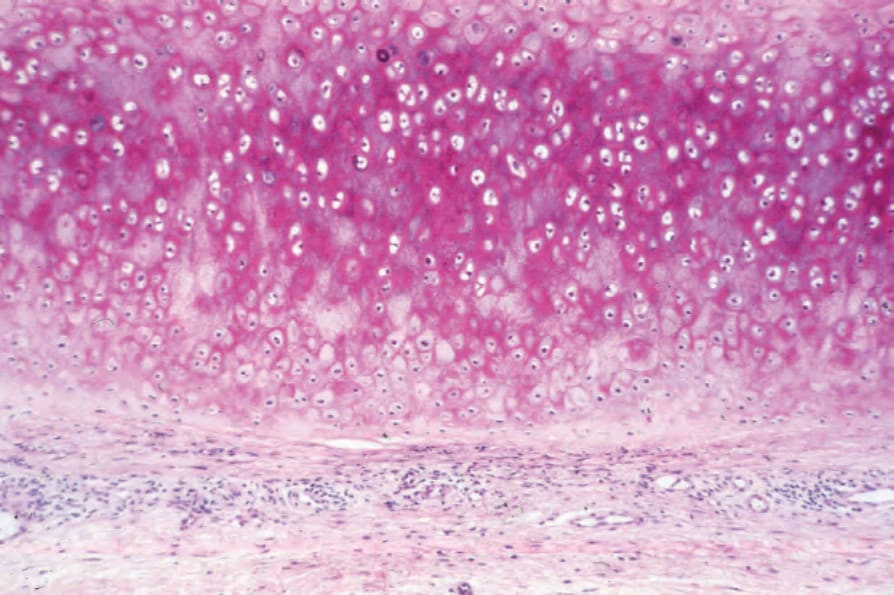
Fig. 17.144 Relapsing polychondritis: a mild chronic inflammatory cell infiltrate is present in the perichondrium.

Table 17.15 Diagnostic criteria for relapsing polychondritis