Eosinophilic fasciitis
Eosinophilic fasciitis
Clinical features The precise nosological status of eosinophilic fasciitis (Schulman syndrome) is uncertain: some authors regard it as a variant of morphea (morphea profunda) but others consider it an entity in its own right.1–4 For the purpose of this text it is classified separately.
Laboratory investigations reveal a raised ESR, peripheral blood eosinophilia, and hypergammaglobulinemia (usually IgG).16 Antinuclear antibodies (speckled and homogeneous), rheumatoid factor and, rarely, anti-nDNA antibodies may be present.11,17 Serum aldolase levels appear to be a useful indicator of disease activity.70 Cytoplasmic-antineutrophilic cytoplasmic antibodies (c-ANCA) have been demonstrated in a patient with recurrent eosinophilic fasciitis.71
Characteristically, eosinophilic fasciitis responds well to corticosteroid therapy – a diagnostic pointer. Spontaneous resolution occurs in some patients. Progression to scleroderma and coexistence with lesions of localized morphea may sometimes occur.7,72–75
Eosinophilic fasciitis occurs equally in males and females, and most patients are in their third to sixth decades.1 Female predominance has been demonstrated in some studies.5,6 Pediatric disease has, however, also been documented.7–10 White Caucasians are predominantly affected.11 Rare co-occurrence in siblings and among family members suggests the possible link with distinct HLA profiles, like HLA-A2.12
The clinical features of painful, tender swelling, stiffness, and sclerodermiform induration affect (in decreasing order of frequency) the forearms, upper arms and lower legs, thighs, hands, trunk, neck, and feet.1,11 The skin changes are often bilateral and symmetrical. The face and fingers are only rarely affected. Localized involvement of a limb has rarely been documented.13 Unilateral presentation is rare.14 Early cutaneous manifestations include pitting edema, peau d’orange or a cobblestone appearance.1,15 At least 50% of patients relate the onset of their illness to an episode of strenuous physical activity.16–19 Patients have a variety of non-specific features including malaise, weakness, fever, and weight loss.1 Raynaud phenomenon is typically absent and the nail fold capillaries are normal – points of distinction from systemic sclerosis.20 Pediatric disease commonly progresses to sclerodermiform cutaneous scarring.7,17,21 Blood eosinophilia and hypergammaglobulinemia have been reported.4,22 Hypogammaglobulinemia is exceptional.23
The criteria for establishing the diagnosis of eosinophilic fasciitis have been proposed recently (see Table 17.11), and require the presence of both major criteria, or alternatively the presence of one major criterion and two minor criteria.76
Pathogenesis and histologic features The etiology and pathogenesis of eosinophilic fasciitis are unknown. The clinical findings of hypergammaglobulinemia, occasional antinuclear antibodies, and positive immunofluorescence suggest a humoral immune mechanism.1 Even in those instances when eosinophilic fasciitis has followed strenuous physical activity, it is unlikely that trauma, on its own, is responsible. There are occasional reports of possible drug toxicity following, for example, antituberculous therapy, phenytoin, subcutaneous heparin and fosinopril, simvastatin, atorvastatin, infliximab (tumor necrosis factor inhibitor), pembrolizumab, and an eosinophilic fasciitis-like picture sometimes constitutes part of the eosinophilia–myalgia syndrome (see below).20,77–82 The disease has also followed exposure to trichloroethylene, radiotherapy, subcutaneous injection of phytonadione, and after the initiation of dialysis.83–86 An exceptional case of unilateral eosinophilic fasciitis following influenza vaccination has recently been reported.87
Extracutaneous involvement is becoming increasingly recognized.24 Patients may develop arthralgia and synovitis. Inflammatory arthritis (predominantly involving the hands, wrists, and knees) and carpal tunnel syndrome occur in about 25% of patients.1,17,20,21,25 Contractures develop in up to 75% of patients and particularly affect the shoulders, elbows, wrists, hands, and knees.12,26 They are the consequence of induration and sclerosis of the subcutaneous tissue and underlying fascia.12,26 Subclinical myositis is common.1 Posterior ischemic optic neuropathy has also been described.27 Clinically significant systemic features are rare, but have included esophageal dysmotility, pericardial and pleural effusions, and lung (restrictive lung disease) and kidney involvement.1,28,29
B. burgdorferi has been associated with some cases of eosinophilic fasciitis (Fig. 17.121).88–90 However, positive serology for B. burgdorferi in
A number of other associations have recently been documented, including aplastic anemia, polycythemia rubra vera, thrombocytopenia, hemolytic anemia, monoclonal gammopathy, X-linked agammaglobulinemia, combined immunodeficiency, paroxysmal nocturnal hemoglobulinuria, abnormal circulating T-cell clone, peripheral T-cell lymphoma, cutaneous T-cell lymphoma, acute myeloid leukemia, myeloblastic leukemia, chronic eosinophylic leukemia, Hodgkin disease, various lymphomas, multiple
Major criteria
- Swelling, induration, and thickening of the skin and subcutaneous
tissues is symmetrical or non-symmetrical, diffuse (extremities, trunk, and abdomen) or localized (extremities)
2. Fascial thickening with accumulation of lymphocytes and macrophages
with or without eosinophilic infiltration (determined by full-thickness wedge biopsy of clinically affected skin)
Minor criteria
- Eosinophilia > 0.5×109/L
- Hypergammaglobulinemia > 1.5 g/L
- Muscle weakness and/or elevated aldolase levels
- Groove sign and/or peau d’orange
- Hyperintense fascia on MR T2-weighted images
After Pinal-Fernandez I., Selva-O’Callaghan A., Grau J.M. (2014) Autoimmun. Rev. 13, 379–382.
815 Eosinophilic fasciitis
patients with eosinophilic fasciitis without direct confirmation of the presence of the organism by polymerase chain reaction has been regarded insufficient for establishing a pathogenetic link.91
In some patients, elevation of serum IL-2, -5, and -10, TGF-β1, tissue inhibitor of metalloproteinase-1, manganese superoxide dismutase, interferon gamma (IFN-γ), and leukemia inhibitory factor has been documented.10,92–94 The increase in IL-5 and -10 possibly leads to eosinophilia and immune globulin overexpression.92 Eosinophils have been shown to stimulate DNA synthesis and matrix production in dermal fibroblasts, leading to increased collagen deposition.95
Immunofluorescence has revealed deposition of IgM at the dermal– epidermal junction, immunoglobulin and complement around blood vessels in the deep dermis, and IgG and complement in the deep fascia and skeletal muscle.11,17,96
The pathology of eosinophilic fasciitis predominantly affects the deep subcutaneous fat and fascia and therefore a substantial incisional biopsy is necessary for diagnosis. The epidermis, papillary dermis, and superficial adnexal structures are usually unaffected.11 A mild chronic inflammatory cell infiltrate consisting of lymphocytes, plasma cells, histiocytes, and variable numbers of eosinophils may be present in the deeper reticular dermis, which is also often fibrosed with atrophy of sweat glands.96 Immunophenotyping of mononuclear inflammatory cell infiltrate demonstrated predominancy of macrophages and CD8+ lymphocytes.97 Occasionally, the dermal changes are indistinguishable from morphea.17
The most dramatic changes are found in the superficial fascia, which is markedly thickened, fibrosed, and sclerotic, and in the acute stages may show focal fibrinoid necrosis and/or myxoid degenerative changes due to excessive glycosaminoglycan deposition (Figs 17.122 and 17.123).1,96 A chronic inflammatory cell infiltrate is present within the fascia in both a diffuse distribution and centered around blood vessels (Fig. 17.124).73 Primary vascular lesions, however, are not a feature. Tissue eosinophilia is focal and often transitory. Its absence in no way precludes the diagnosis. Lymphoid follicles, sometimes with germinal centers, are also occasionally evident.96 The inflammatory changes usually extend into the septa of the subcutaneous fat and fibrosis may result in fat entrapment.16 There may also be superficial infiltration by inflammatory cells into the underlying skeletal muscle, which occasionally shows focal necrosis, degeneration, and foci of regeneration.1,96,98
Differential diagnosis While there is obvious overlap with morphea, the diffuse nature of the induration clinically, the high peripheral eosinophilia, and history of preceding strenuous exercise, combined with the usually less severe dermal changes
816 Idiopathic connective tissue disorders
and preservation of the skin appendages on histology, commonly serve to distinguish the two disorders.
Sclerodermoid and eosinophilic fasciitis-like syndromes have been described as features of the toxic oil and L-tryptophan-related eosinophilia– myalgia syndromes.99–104
• The toxic oil syndrome arose as a consequence of contaminated rapeseed oil and presented in Spain in 1981.104,105 Patients developed myopathy, peripheral neuropathy, and arthralgia in addition to morphea-like skin induration affecting the face, trunk, and extremities.104 Xerostomia was common and Raynaud phenomenon was not infrequent.
• The eosinophilia–myalgia syndrome is due to contaminated (Peak E) commercial L-tryptophan. Patients develop a wide variety of clinical and laboratory abnormalities involving the skin, muscles, nerves, fascia, and lungs.101 Acute cutaneous involvement is most commonly seen as a non-specific erythematous macular eruption affecting the trunk and extremities.101 Chronic lesions include edema of the extremities followed by the development of sclerodermiform and/or eosinophilic fasciitis-like features.99
and it is not that rare for the skin eruption to precede the onset of muscle involvement by more than 2 years.8,9 The proposed concept of amyopathic dermatomyositis or dermatomyositis sine myositis has been the source of much controversy.10–15 In addition to a prolonged follow-up, adequate electromyographic studies and muscle biopsy are mandatory before accepting that the patient has only cutaneous lesions.15,16 Only 5% or less of cases of dermatomyositis can be classified as the amyopathic variant after long-term follow-up.14 For the diagnosis of amyopathic dermatomyositis to be made, it has been suggested that there should be absence of clinical or laboratory signs of muscle disease for at least 2 years after the onset of skin disease.17 The clinical cutaneous signs of amyopathic dermatomyositis are identical to those seen in classic dermatomyositis. Patients with amyopathic dermatomyositis, like those with dermatomyositis/polymyositis, are at increased risk of developing severe interstitial fibrosis of the lung and associated malignancies.16
Variable histologic features have been documented, presumably reflecting different stages of evolution. In some patients the most conspicuous changes have included fibrosis involving the papillary dermis, periappendageal connective tissue sheath, and subcutaneous fat.100 An inflammatory cell infiltrate composed of lymphocytes, histiocytes, plasma cells, and variable numbers of eosinophils is present in the dermis, subcutaneous fat, and fascia.101,102 Mast cells are sometimes conspicuous.102 Late stages are characterized by hyaline sclerosis involving the dermis through to the subcutaneous fat.102 Additional features that have been documented include dermal edema with lymphangiectasia and heavy mucin deposition in both the dermis and fascia.100 The histologic features overlap between morphea/systemic sclerosis and eosinophilic fasciitis.
Dermatomyositis/polymyositis is associated with severe morbidity and high mortality, the latter particularly reflecting cardiac and pulmonary involvement.
The cutaneous features are usually quite distinctive.18 Commonly, the patient presents with periorbital edema and a reddish-violet discoloration, often termed heliotrope erythema (heliotrope flower) (Figs 17.125 and 17.126). The upper eyelids are most often affected and the eruption is typically symmetrical.19 Nevertheless, unilateral presentation of heliotrope rash has also been reported and can represent a diagnostic pitfall.20 Heliotrope erythema is usually associated with a lupus-like erythema, which involves the rest of the face and spreads to the neck, upper trunk, and extensor surfaces of the limbs and dorsal aspects of the hands and fingers (Fig. 17.127). It is associated with a slight scale. Exceptionally, generalized subcutaneous edema may develop.21
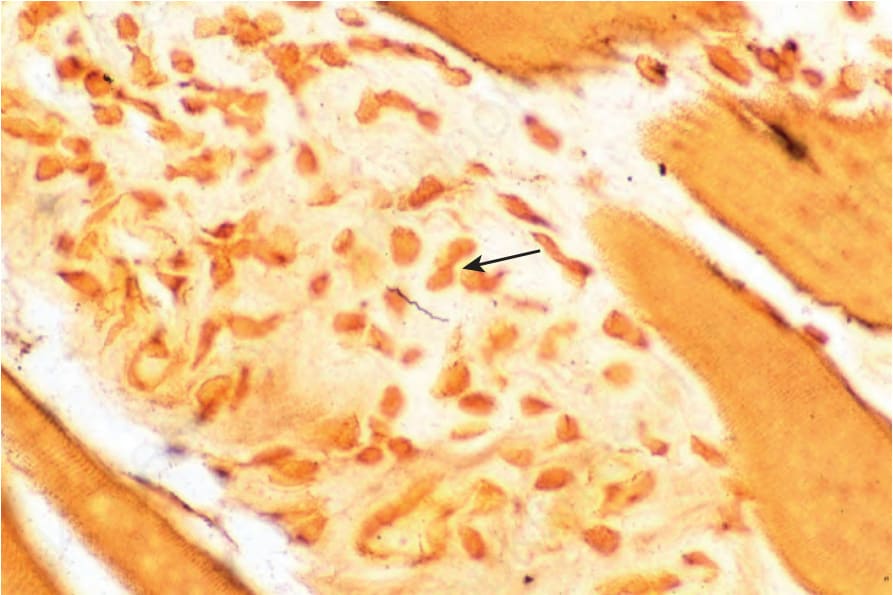
Fig. 17.121 Eosinophilic fasciitis: a spirochete is present in the center of the field (Dieterle stain) (arrow).
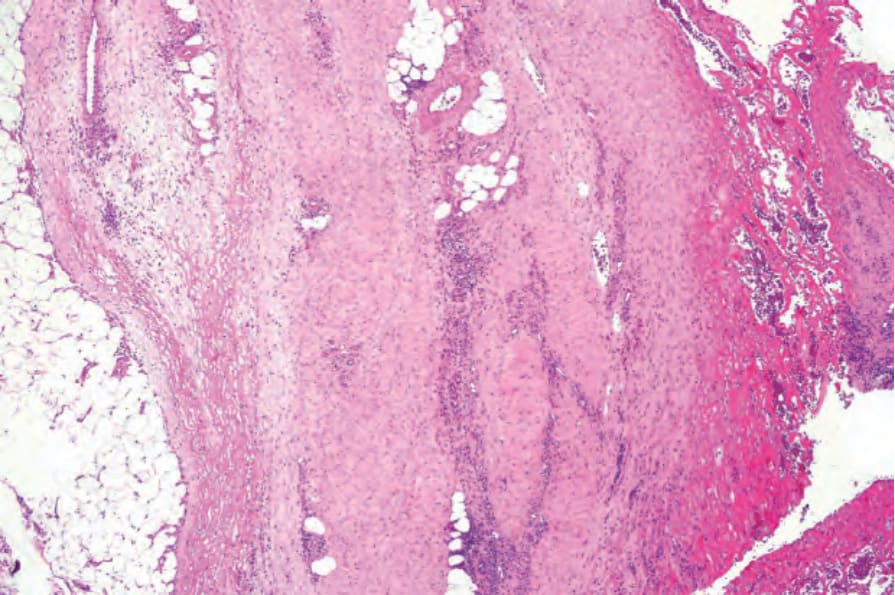
Fig. 17.122 Eosinophilic fasciitis: the fascia is thickened and there is marked fibrin deposition.
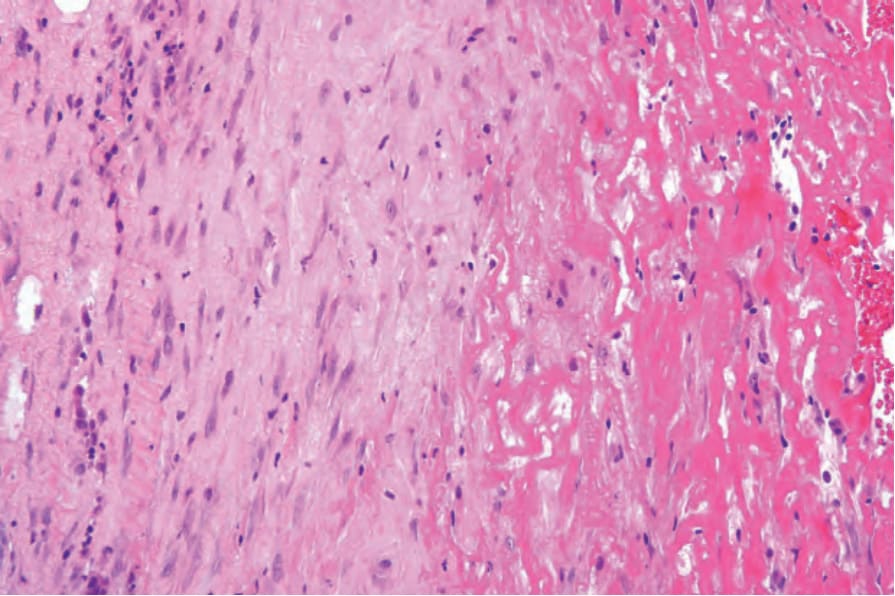
Fig. 17.123 Eosinophilic fasciitis: high-power view.
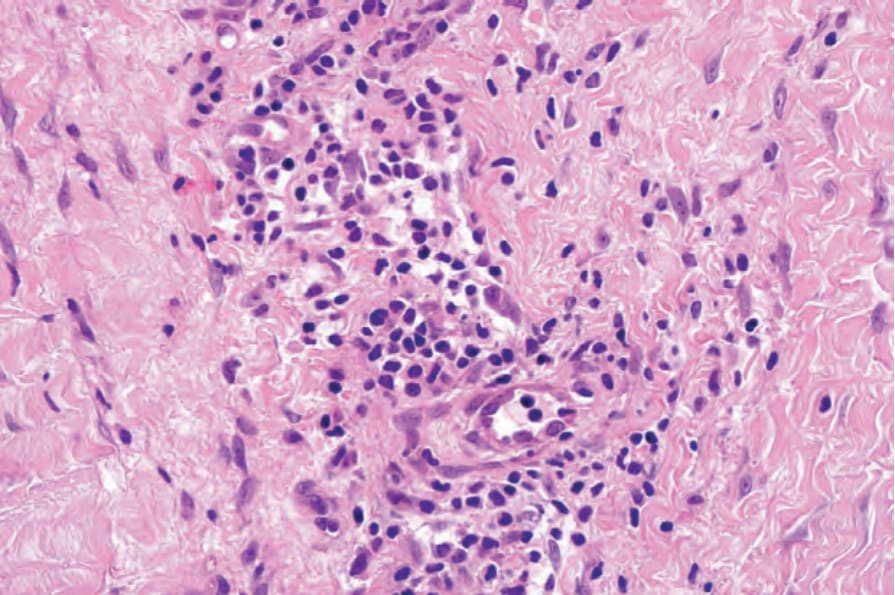
Fig. 17.124 Eosinophilic fasciitis: the infiltrate consists of lymphocytes with only one or two eosinophils.
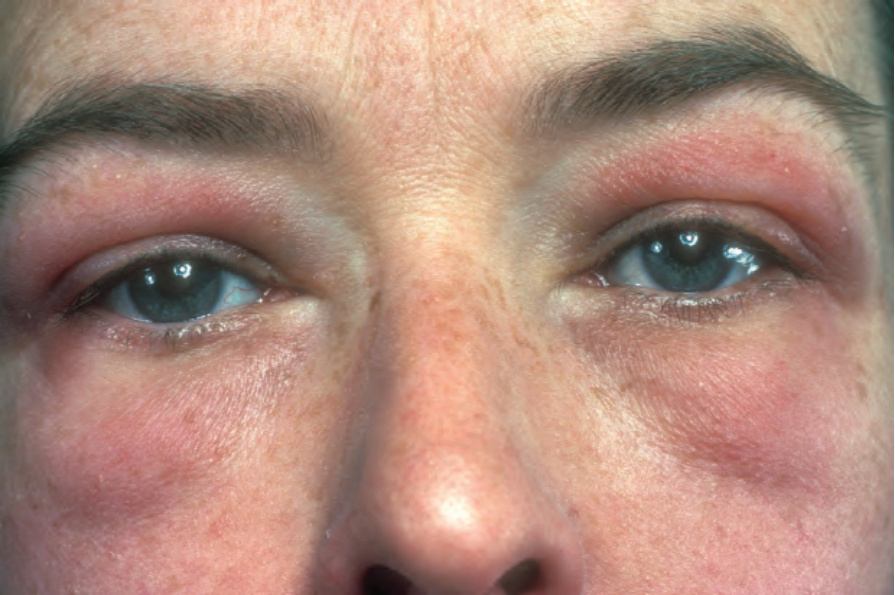
Fig. 17.125 Dermatomyositis: note the characteristic red-mauve discoloration around the eyes. There is also spread onto the cheeks. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
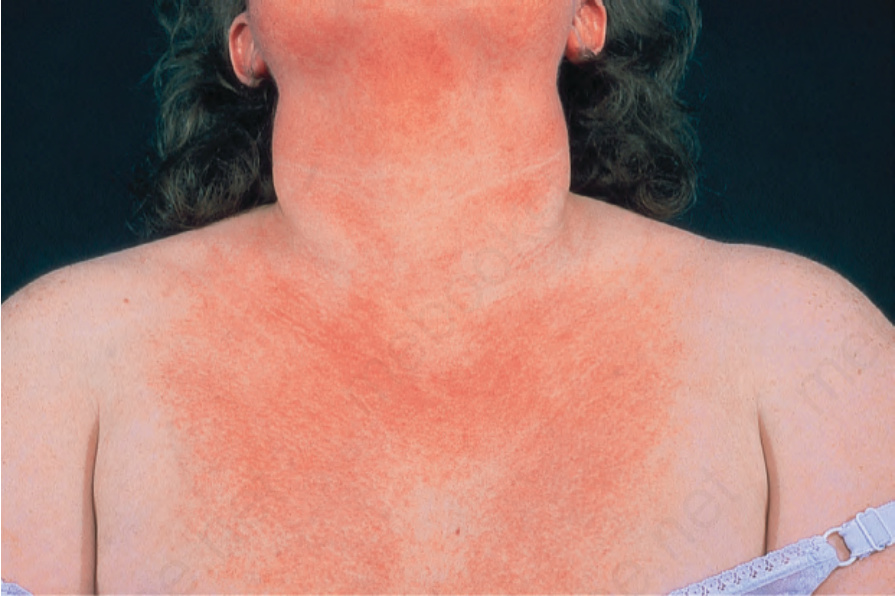
Fig. 17.127 Dermatomyositis: note the erythema and slight scale on this patient’s chest. By courtesy of the Institute of Dermatology, London, UK.

Table 17.11 Proposed criteria for the diagnosis of eosinophilic fasciitis