Localized scleroderma (morphea)
Localized scleroderma (morphea)
Localized scleroderma (morphea) constitutes a group of diseases characterized by thickening or sclerosis of the dermis with loss of subcutaneous fat, sometimes with involvement of the underlying skeletal muscle.1–6 The incidence of localized scleroderma has been estimated to be at 27 patients per 1 000 000.7–9 In contrast, the juvenile variant of localized scleroderma has an incidence of 3.4 patients per 1 000 000 children.9 There is predilection for children and young adults, with the peak incidence between 20 and 40 years, and females being up to six times more frequently affected.7,10–12 Rare congenital cases have been documented.13–15 About 15% of cases develop before the age of 10 years.16 Localized scleroderma is not usually associated with severe systemic symptoms or Raynaud phenomenon, is often self-limited, and in general has a good prognosis, although the linear variant in particular may be very disabling and often disfiguring, especially in children.2 The linear and deep variants can be associated with arthralgias, synovitis, uveitis, and joint contractures.17 A large study of patients with morphea
Morphea usually develops slowly and the onset may manifest as erythema and edema. An established lesion is typically circumscribed, ivory or white in color, and densely sclerotic (Fig. 17.103).2 A characteristic feature is the presence of a violaceous border, an indicator of disease activity (Figs 17.104 and 17.105).2 As the lesion subsides, atrophy, loss of hair and sebaceous glands, and variable hypo- and hyperpigmentation become evident (Fig. 17.106).34 Vesicles, bullae, purpura, and telangiectasia may rarely be present, particularly in the generalized variant.1,37 Tense bullae, due to subepidermal edema, are a rare manifestation that has been described in morphea, including the profunda variant.38 They are thought to develop as a consequence of both trauma and lymphatic obstruction. The latter is suggested by the finding of lymphatic dilatation in 77% of biopsies from patients with this variant of morphea.39 It has also been suggested that this type of morphea may be related to release of major basic protein from eosinophils.39
808 Idiopathic connective tissue disorders
The plaque-form of morphea usually consists of multiple, round or oval, sometimes pruritic, 2–15-cm diameter lesions, which are usually bilateral and asymmetrical in distribution (Fig. 17.107). Lesions occur (in decreasing order of frequency) on the thorax and neck, the lower extremities, the upper extremities, and the face; the axillae, umbilical region, perineum, and perianal area are usually spared (Fig. 17.108).
The so-called linear atrophoderma of Moulin, which presents with bandlike lesions following Blaschko lines, is likely to represent a variant of linear morphea.40
Linear morphea is usually solitary and unilateral in distribution. Lesions are found (in decreasing order of frequency) on the lower limbs, the upper limbs, the frontoparietal region (e.g., en coup de sabre), and the anterior thorax (Fig. 17.109). Linear lesions may involve both the upper and lower extremities simultaneously and, on occasion, plaque-type morphea is also present.2 Although the clinical appearances of linear scleroderma are very similar to those of the plaque-form, lesions tend to show more pigmentary change and the violaceous border is less conspicuous. Linear morphea may affect the underlying skeletal muscle and even bone, giving rise to contractures and deformities. Calcification of skeletal muscle may exceptionally occur.41 An association with melorheostosis (an uncommon mesenchymal dysplasia or Leri disease) has been described.38,39,42 Cases presenting with
809 Localized scleroderma (morphea)
hypertrichosis are also documented.43–45 Occasionally, it follows Blaschko lines.46–51
Frontoparietal linear morphea presents as a densely sclerotic plaque extending from the eyebrow onto the scalp and may be associated with alopecia. Involvement of the cheek, nose, and upper lip has also been documented.34 Progression of the lesion results in the development of a groove and hence the term ‘en coup de sabre’ (Fig. 17.110). Gingival recession has occasionally been documented.52 Familial occurrence is exceptional and bilateral lesions are rare.53–55 A further complication, particularly in children, is the development of facial hemiatrophy (Romberg disease) (Fig. 17.111).2,56 Exceptionally, central nervous system involvement may occur,57 with presentations such as intractable partial seizures, headache, focal
neurological deficit, recurrent myelitis, and cerebral vasculitis.58–60 About 50% of patients additionally display various dental abnormalities.61 Linear morphea has also been described in association with hereditary deficiency of C2.62
Guttate morphea In guttate morphea lesions are multiple, small (2–10 mm), nonindurated, and yellowish-white, and are limited by a delicate lilac border.1,34 Typically, there is no hyperkeratosis or follicular plugging. Coalescence of lesions to form plaques is not uncommon. Guttate morphea most commonly presents on the upper back and shoulders, but the lower back, chest, and abdomen may also be affected.1 There is much clinical (and histological) overlap
810 Idiopathic connective tissue disorders
the deep dermis and subcutaneous fat. It usually affects the scalp, face, trunk, and extremities.1,87–89 An adult-onset variant of disabling pansclerotic morphea has also been reported.90 Disabling pansclerotic morphea is more common in males.91 Patients have tendencies for chronic nonhealing ulceration, most commonly involving legs, followed by upper extremities, trunk, and head.89 Patients may also have arthralgias, contractures that particularly affect the extensor surfaces of the extremities, and osteoporosis.34,87 This exceedingly severe variant of localized scleroderma is unremitting and permanent, and is fortunately very rare. Some patients have had abnormal respiratory function tests and diminished esophageal motility, suggesting overlap with systemic sclerosis.87 Blood eosinophilia is also seen.87 A rare complication of squamous cell carcinoma has been documented.92,93 A further exceptional association is that of hypogammaglobulinemia.94
Associated conditions Localized scleroderma has been associated with a variety of conditions including arthralgia, carpal tunnel syndrome, unilateral Raynaud phenomenon, intermittent abdominal pain, and spina bifida.34,35 Concurrent lichen planus, often in the company of lichen sclerosus, has also been documented.1 A large-perspective German multicenter study of 472 patients with localized scleroderma detected a high prevalence of lichen sclerosus in these patients (5.7%), especially in the anogenital region.95 Other associations include vitiligo, alopecia areata, granuloma annulare, pigmented purpuric dermatosis, psoriasis vulgaris, cutaneous amyloid deposition, lupus anticoagulant, DLE, SCLE, SLE, xanthomatosis, elastosis perforans serpiginosa, B-cell lymphoma, multiple myeloma, carcinoid syndrome, human T-cell lymphoma/lymphotropic virus type 1 infection, chronic hepatitis B and C virus infection, posthepatitis C cirrhosis, primary biliary cirrhosis, Rosai-Dorfman disease, sarcoidosis, necrotizing vasculitis, necrobiotic xanthogranuloma, hepatosplenomegaly, multiple lymphadenopathy, polymyositis, neurofibromatosis type I, port wine stain, and regional inflammatory myopathy.1,2,96–128 A recent analysis of 245 patients with localized scleroderma has found concomitant rheumatic or autoimmune disorder in 17.6%.129 Generalized morphea is the most frequent subtype associated with autoimmune diseases, which is present in 45.9% of patients with this form of the disease.129
between the lesions of guttate morphea and lichen sclerosus and it is worthy of note that the two disorders are frequently seen together.2,63
Generalized morphea Generalized morphea, which most commonly affects the trunk and abdomen, is characterized by widespread large lesions resembling plaque-type morphea.1,64 These may merge and in many patients almost the entire skin surface is involved. Lesions are often symmetrically distributed and frequently exhibit different disease stages.65 Extension to the subcutaneous fat and muscle sometimes results in severe contractures and disabling and disfiguring deformities (Fig. 17.112). Generalized morphea may occasionally prove fatal, for example, due to pneumonia. Rarely, systemic involvement supervenes.2 When generalized morphea and systemic sclerosis coexist, the activity of the generalized morphea may be independent of the lesions of systemic sclerosis.66 An association with porphyria cutanea tarda, eosinophilic fasciitis, and childhood sclerodermatomyositis has been described.67–69 Occurrence with Felty syndrome and after antitetanus vaccination has also been reported.70,71 Unilateral generalized morphea has been documented.72,73 A patient with this form of the disease developed multiple acral adult myofibromas.74 Multiple squamous cell carcinomas of the skin developing in the background of generalized morphea are an exceptional finding.75
Pathogenesis and histologic features Localized scleroderma in characterized by excess deposition of collagen type I by intrinsic activation of TGF-β signaling.130 Nevertheless, the exact etiology and pathogenesis of localized scleroderma are unknown. Theories of causation include trauma, hormonal influences, and familial aspects.34,131,132 Thus localized scleroderma may present or worsen during pregnancy, the menarche, or the menopause. The condition has also been described following chickenpox and measles.1,133 An infectious etiology has received some support with the identification by immunohistochemistry, silver stains, and polymerase chain reaction (PCR) of Borrelia burgdorferi in biopsies of lesional skin combined with the presence of elevated antibody levels.134–141 Lymphoproliferative responses to this organism have also been reported in patients with morphea.142 Most studies, however, have cast doubt on the association between morphea and B. burgdorferi.143–152 It has also been shown that false-positive tests for B. burgdorferi with indirect immunofluorescence and even enzyme-linked immunosorbent assay (ELISA) are not uncommon.153 It is therefore more likely that B. burgdorferi is not etiologically linked to localized morphea. A further possibility is that only certain subspecies of Borrelia are capable of inducing the disease.154,155 However, this theory has not been substantiated by different studies from the same country.155
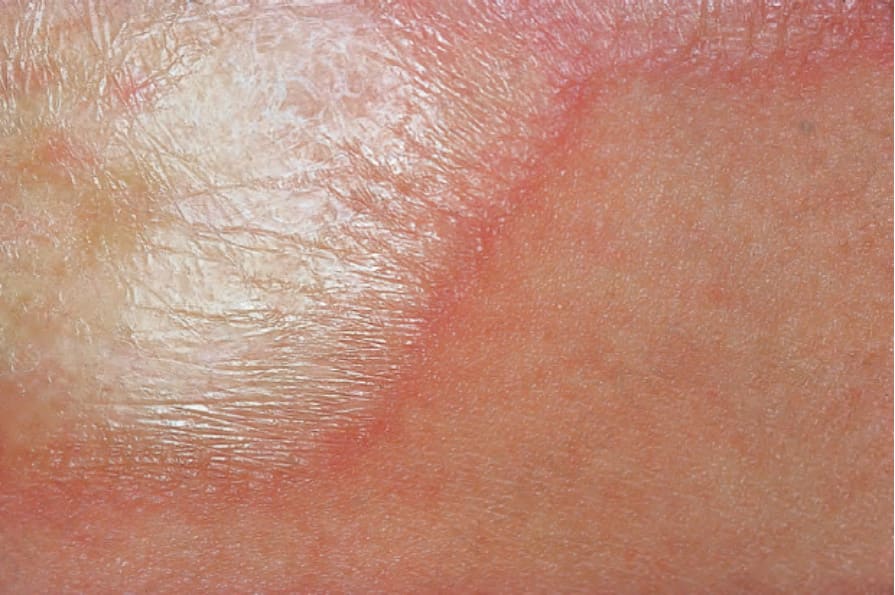
Fig. 17.103 Morphea: characteristic white sclerotic plaque. By courtesy of the Institute of Dermatology, London, UK.
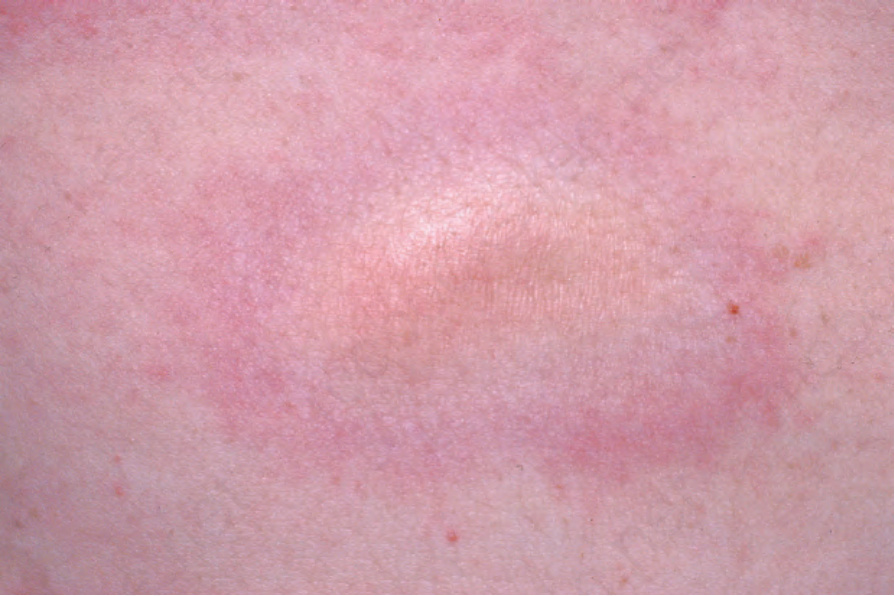
Fig. 17.104 Morphea: in this example the violaceous border is apparent. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
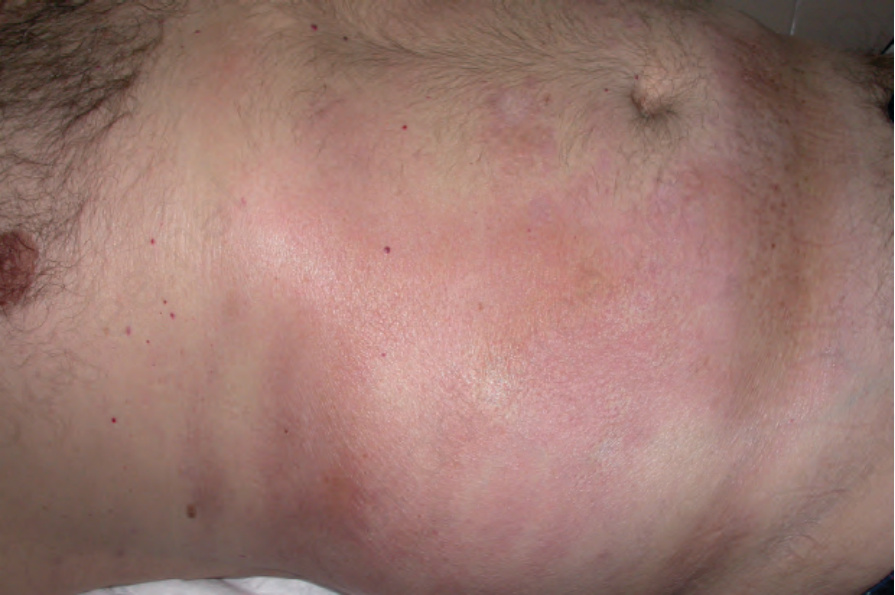
Fig. 17.105 Morphea: multiple lesions are present on the abdomen. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
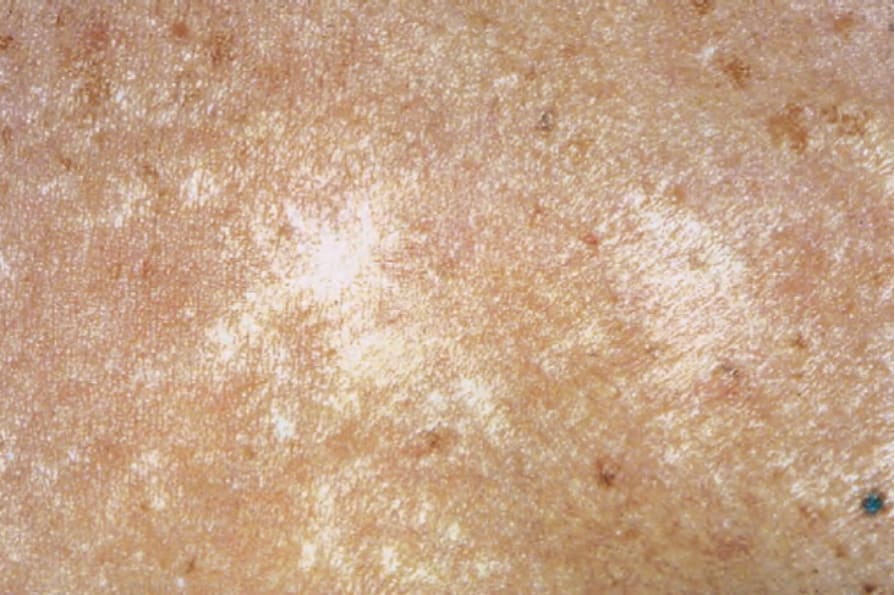
Fig. 17.106 Morphea: atrophic lesions showing variable hypo- and hyperpigmentation. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
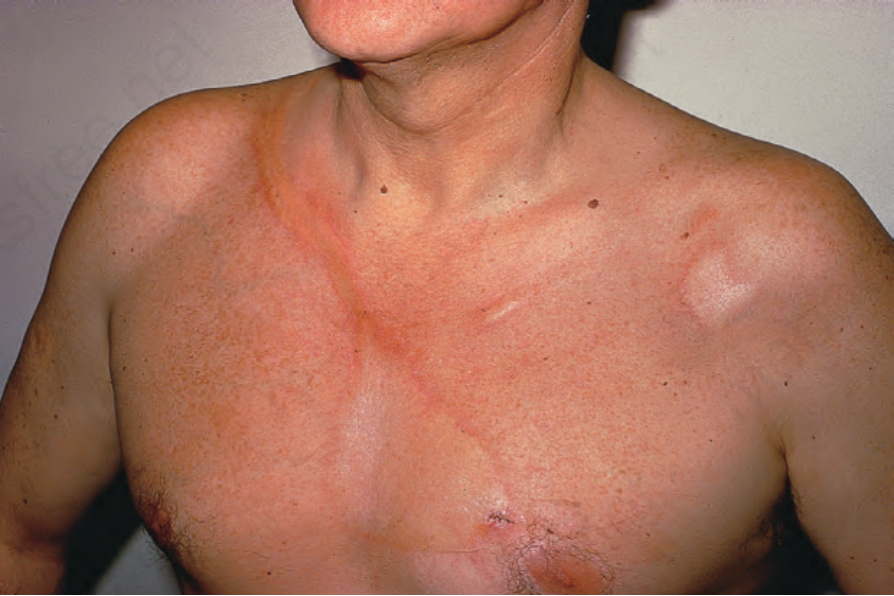
Fig. 17.107 Morphea: multiple asymmetrical lesions are present. Note the characteristic en coup de sabre. By courtesy of D. McGibbon, MD, St Thomas’ Hospital, London, UK.
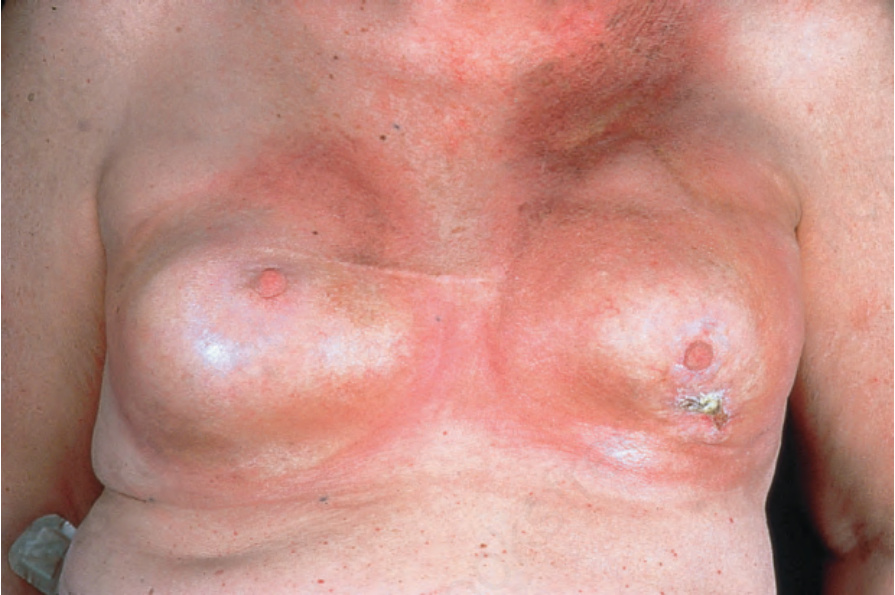
Fig. 17.108 Morphea: extensive lesions can be very disfiguring, as in this patient showing bilateral breast involvement. By courtesy of the Institute of Dermatology, London, UK.
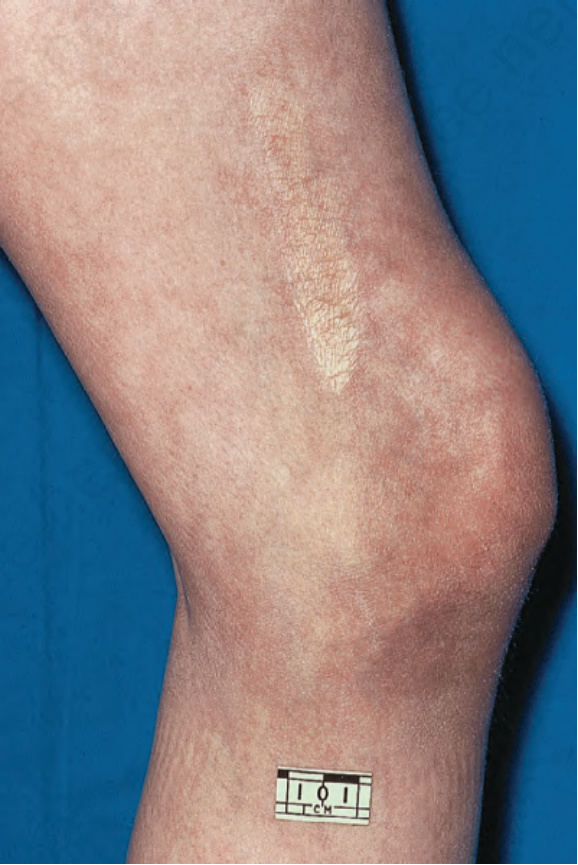
Fig. 17.109 Linear morphea: atrophic lesion on the thigh. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
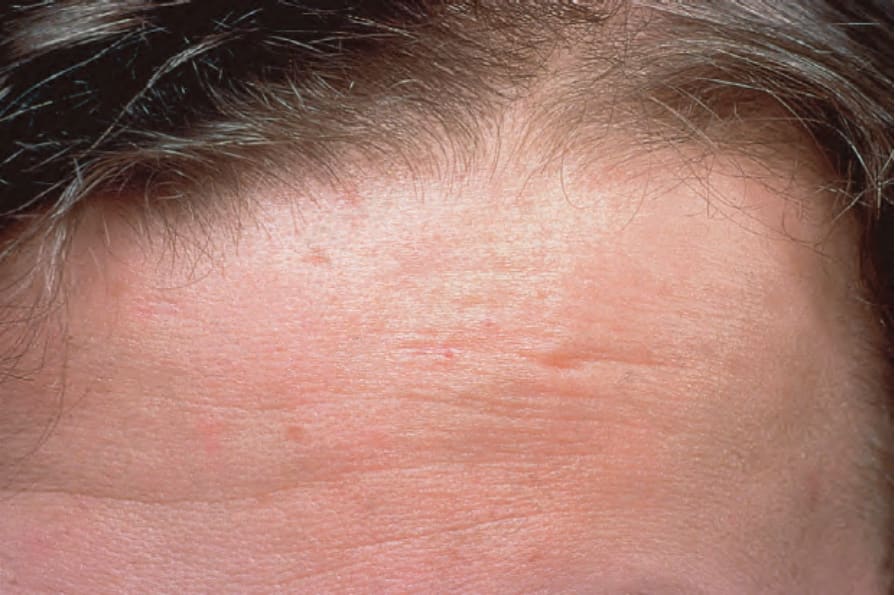
Fig. 17.110 Linear morphea: en coup de sabre. By courtesy of the Institute of Dermatology, London, UK.
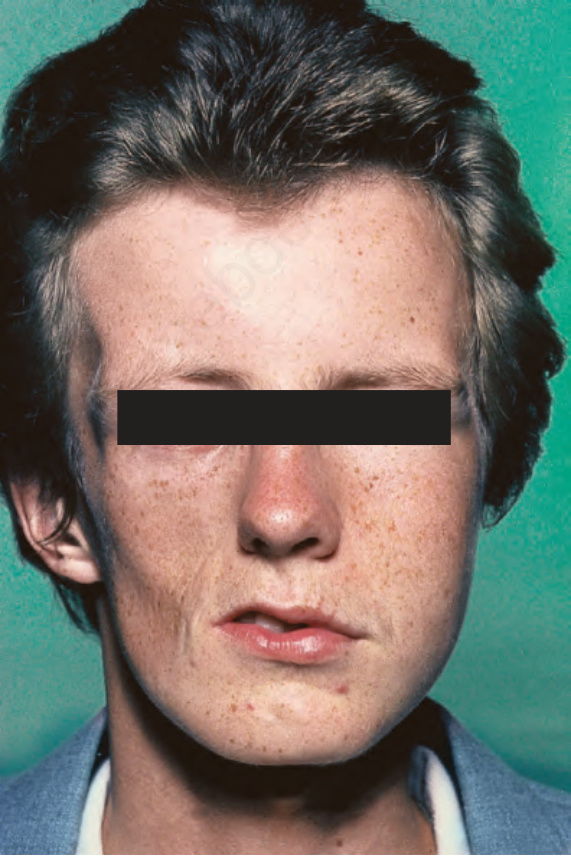
Fig. 17.111 Linear morphea: severe facial hemiatrophy. By courtesy of D. McGibbon, MD, St Thomas’ Hospital, London, UK.
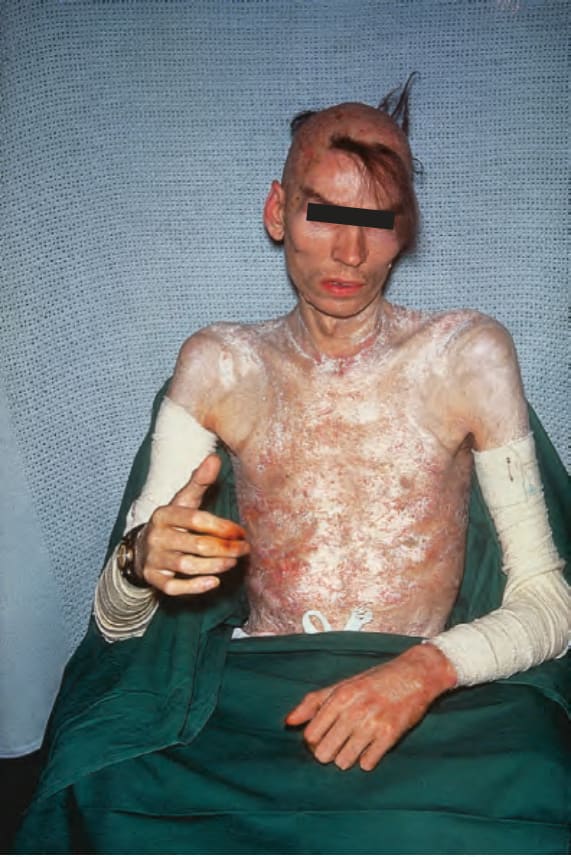
Fig. 17.112 Generalized morphea: a very advanced extreme example showing almost complete involvement of the skin, hair loss, and contractures. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.

Fig. 17.115 Morphea: the changes are highlighted with this Masson trichrome stained section. In this example, the papillary dermis is involved.
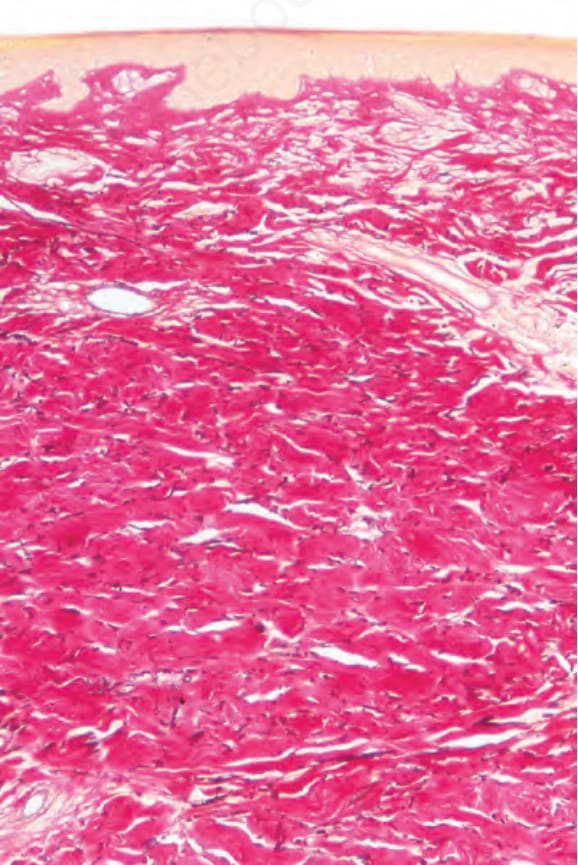
Fig. 17.116 Morphea: this is a chronic lesion. There is loss of elastic tissue (van Gieson).
Subcutaneous scleroderma Subcutaneous scleroderma (morphea profunda, nodular scleroderma, keloidal scleroderma) presents clinically as nodular or keloid-like lesions.34,38,76–82 Association with systemic sclerosis can be present.81,83 The abdomen, sacral region, and the extremities are affected most commonly.2 Osteoma cutis can rarely develop in subcutaneous scleroderma.84 Two cases of morphea profunda, as well as an atrophic variant of morphea profunda mimicking localized lipoatrophy, have been reported at the site of previous intramuscular vaccination.85,86
Disabling pansclerotic morphea of children Disabling pansclerotic morphea of children is a particularly aggressive and mutilating variant, which involves fascia, muscle, and bone in addition to
The occasional simultaneous occurrence of localized scleroderma and systemic sclerosis has led some authors to postulate a shared pathogenetic mechanism.35,156 In both conditions increased serum levels of procollagen type I carboxyterminal propeptide have been reported.157 Similarly, the presence of localized scleroderma in both discoid and systemic lupus erythematosus and dermatomyositis has been cited as additional evidence for an immunological basis.1,158–160 It is also of interest that the clinical appearances and histology of late GVHD are very similar to those of scleroderma. Increased expression of connective tissue growth factor has been detected in
sclerotic fibroblasts of nodular scleroderma by immunohistochemistry and in situ hybridization.83
Antinuclear antibodies may be detected in approximately 70% of patients with morphea.161 Homogeneous, nucleolar, and speckled patterns have all been recognized, but the first is the most common variant.162 Rheumatoid factor, anti-dsDNA, anticentromere, and anti-Scl-70 antibodies have also been documented but are rare.64 Anti-ssDNA antibodies are present in 38% to 75% of patients, are frequently of the IgM subclass, and are found more often in linear and generalized morphea than in the plaque form.64,163 Antihistone antibodies have been reported in up to 50% of cases.164 Most antibodies are more commonly seen in patients with active or widespread disease.160 Antinuclear antibodies are frequent in children with localized scleroderma and often have specificity for denatured DNA and for high mobility group proteins.165 Anti-Cu/Zn superoxide dismutase antibodies are present in the serum of 89% of patients with localized scleroderma, and 100% of patients with the generalized variant.166 Anti-DNA topoisomerase IIα antibodies have been detected in 76% of patients with localized scleroderma, and 85% of patients with the generalized variant.167 Increased serum levels of ICAM-1 have also been reported, particularly in patients with prominent involvement.168 Peripheral eosinophilia is sometimes a feature, particularly in the pansclerotic morpheic variant.1,87
811 Localized scleroderma (morphea)
Direct immunofluorescence studies have demonstrated immunoglobulin (usually IgM) and complement at the basement membrane region and around the dermal vasculature in about 35% of patients.1 Generalized morphea is more often positive than the plaque and linear variants. Immunohistochemical studies of established lesions reveal increase in the number of factor XIIIa+ cells and decrease in the number of CD34+ cells.169–171
The histologic features of localized scleroderma involve both the dermis and subcutaneous fat; a deep incisional biopsy is therefore indicated if scleroderma is suspected.64,163 Biopsies from early lesions often show very subtle histologic findings and are frequently non-specific. The histologic diagnosis may be more difficult to establish in biopsies of lesions from guttate morphea as the changes tend to be more focal and superficial. In an established indurated plaque, the epidermis is usually normal or occasionally flattened. Mucin deposition is not usually a feature but may be occasionally present. Abundant mucin throughout the reticular dermis has been described exceptionally in nodular morphea.51,172 The papillary dermis either appears unaffected or shows a rather homogenized change (Fig. 17.113). The most striking features are seen in the reticular dermis, where the collagen bundles are swollen, intensely eosinophilic, and orientated parallel to the surface (Figs 17.114–17.116). There is also involvement of the septa of the subcutaneous fat; this is associated with atrophy of the adipocytes and subsequent fibrosis, resulting in an apparent increase in thickness of the dermis.1 Hair follicles and sebaceous glands may be atrophic or absent and the eccrine ducts often appear compressed within the densely sclerotic dermis. Due to fibrous replacement of the subcutaneous fat, the eccrine glands appear to be situated abnormally high within the dermis rather than at the dermosubcuticular interface. In rare cases only the superficial reticular dermis is affected.173
An important feature of localized scleroderma, especially in the early stages, is a dense, chronic inflammatory cell infiltrate of lymphocytes, histiocytes, and plasma cells; some authors believe this to be the initial feature (Fig. 17.117).64 Eosinophils can also be present. The infiltrate may surround blood vessels and appendages and tends to be particularly conspicuous in the dermis in addition to the subcutaneous fat (Fig. 17.118). A recent histologic study in patients with ‘en coup de sabre’ localized scleroderma revealed the presence of vacuolar degeneration of keratinocytes in the epidermis and follicular epithelium to be a consistent finding in early and active lesions.174 In addition, perineural inflammatory cell infiltrate in a concentric pattern with plasma cells neurotropism is also commonly observed.175,176 In the linear and generalized variants in particular, the inflammatory changes may affect the underlying skeletal muscle.
In the late stages, dermal sclerosis is still evident, but the dermis appears thinned due to concomitant atrophy.2 Vascular changes similar to those described for systemic sclerosis may be evident and consist of thickening of the walls of small blood vessels. Vasculitis is not a feature. Calcinosis cutis
is occasionally seen and neuritis similar to that seen in indeterminate leprosy has also been documented.177,178
In lesions of deep morphea the infiltrate is much more prominent and is located predominantly in the junction between the dermis and subcutaneous tissue with extension into the subcutaneous tissue. The infiltrate and the sclerotic collagen have a more nodular distribution.
812 Idiopathic connective tissue disorders
Several patterns have been described in bullous morphea.39,179,180 The most common is that of prominent superficial edema with prominent lymphatic dilatation.39 A further pattern is one identical to that seen in lichen sclerosus.180 It is worth remembering that autoimmune blistering diseases, such as epidermolysis bullosa acquisita, may occur concomitantly with
morphea, and immunofluorescence may be indicated in cases in which a subepidermal blister is present.179
In addition to the features described above, generalized morphea and disabling pansclerotic morphea of childhood may show a lymphocytic and hyaline panniculitis with lymphoid follicle formation reminiscent of lupus
813 Atrophoderma of Pasini and Pierini
profundus.64,87 Eccrine squamous syringometaplasia and syringomatous hyperplasia have been described in linear scleroderma.181
Differential diagnosis The lesions of localized scleroderma may be histologically indistinguishable from those of systemic sclerosis, but the inflammatory cell infiltrate tends to be more pronounced in the former, at least in the early stages. Also, involvement of the papillary dermis may be a feature in some cases of localized scleroderma.182 In addition, a more diffuse dermal and less prominent subcutaneous sclerosis coupled with more intense inflammation, including perineural inflammation, are features more often seen in localized scleroderma.175
Other diseases that enter the differential diagnosis include late porphyria cutanea tarda and chronic GVHD. Adequate clinical information will resolve most diagnostic dilemmas, but where doubt exists, the presence of PAS-positive thickened dermal vessels is indicative of porphyria, whereas epidermal lichenoid features with cytoid body formation strongly support the diagnosis of chronic GVHD.
predilection for females (5 : 1).6 A congenital variant of atrophoderma of Pasini and Pierini has also been described.7–10
The typical lesion is a gray-brown or violaceous, atrophic, round to oval, depressed nonindurated macule with a ‘cliff-drop’ border (Fig. 17.120).3 The distribution is usually bilateral and symmetrical.6 Widespread unilateral involvement is rare.11 While previous studies demonstrated the lower back as the most commonly affected site,6 recent analysis of 16 patients revealed predominance of lesions on lower extremities (62.5%), followed by upper extremities and trunk.12 Lesions may also be found on the chest, arms, and abdomen. A zosteriform distribution of the lesions has occasionally been documented.13,14 The lesions are frequently hypopigmented.12 A linear variant of atrophoderma (of Moulin) with the distribution of the lesions following Blaschko lines has been regarded a variant of atrophoderma of Pasini and Pierini.15–18
The relationship between localized scleroderma, particularly the guttate variety, and lichen sclerosus has been the source of considerable controversy. However, basal cell liquefactive degeneration with a lichenoid inflammatory cell infiltrate is not a feature of morphea, and sclerosis of the reticular dermis and subcutaneous fat with atrophy or loss of appendage structures is not seen in lichen sclerosus (Fig. 17.119).63
Marked dermal sclerosis may also be a feature of atrophie blanche and chronic radiation dermatitis. Vascular changes, including thromboses, purpura, and hemosiderosis, however, are conspicuous in the former, while bizarre fibroblasts, elastosis, and endarteritis obliterans are characteristic of the latter.
Atrophoderma of Pasini and Pierini may coexist with lichen sclerosus and morphea, and progression to systemic sclerosis has been documented.13,19–22 In contrast to localized scleroderma, it lacks the violaceous border, is primarily atrophic rather than indurated, and tends to great chronicity, lesions often being present for decades rather than resolving after a few years, as is often a feature of morphea.3
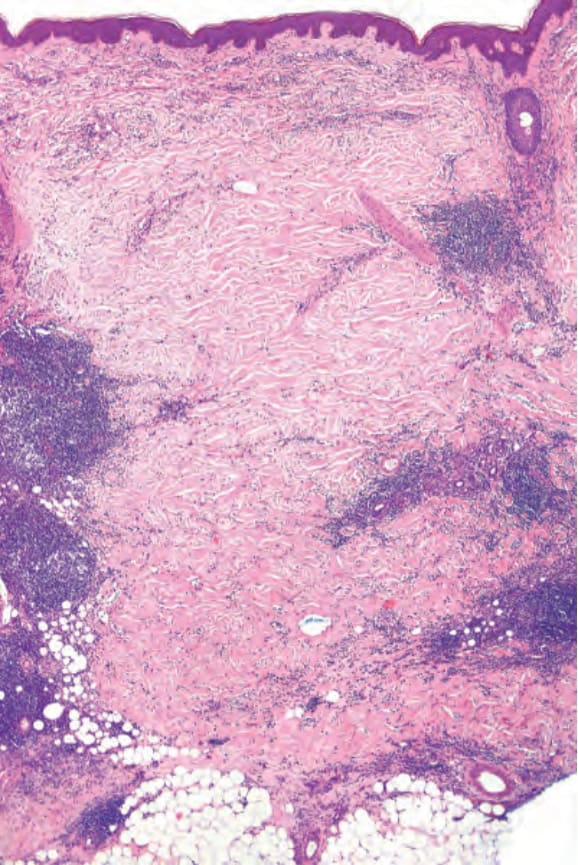
Fig. 17.113 Morphea: the dermis is thickened by dense collagen bundles. Note the heavy perivascular infiltrate.
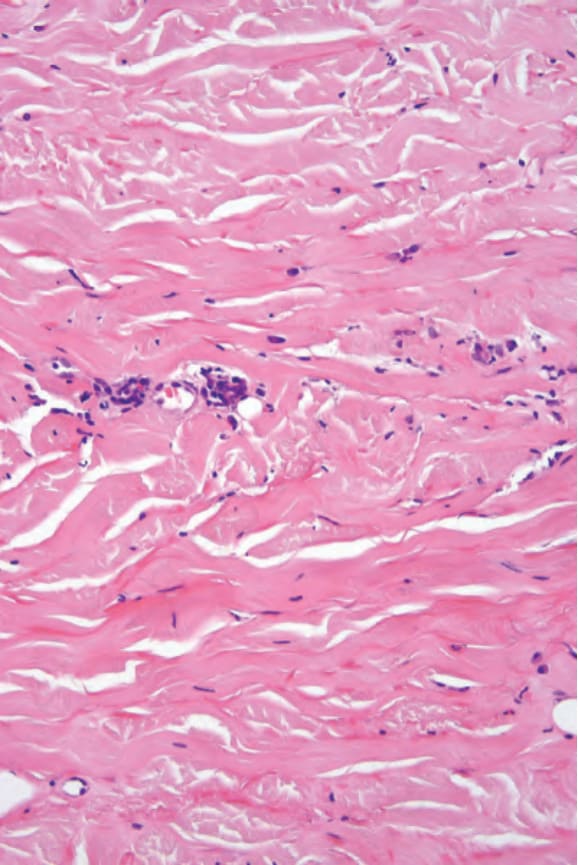
Fig. 17.114 Morphea: the collagen fibers are eosinophilic and swollen.
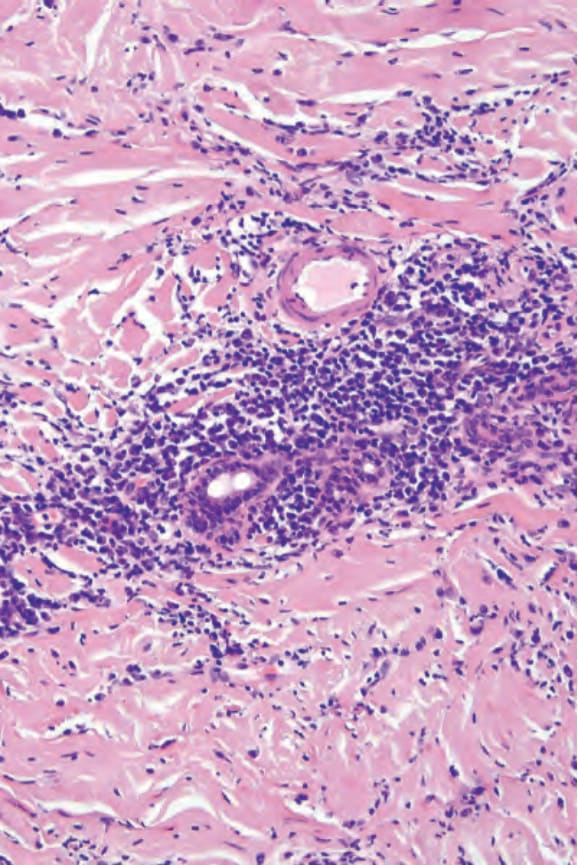
Fig. 17.117 Morphea: a perivascular chronic inflammatory cell infiltrate is usually present, particularly in early lesions.
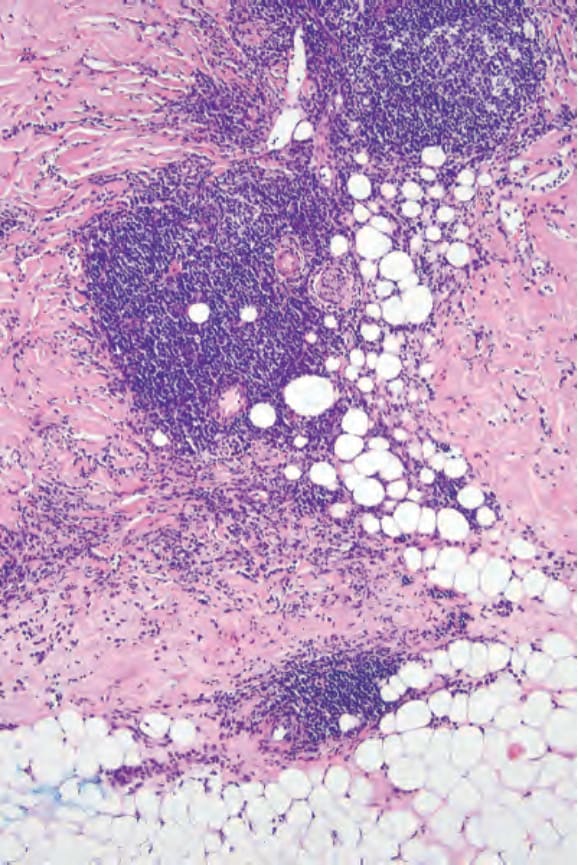
Fig. 17.118 Morphea: the infiltrate often involves the subcutaneous fat.
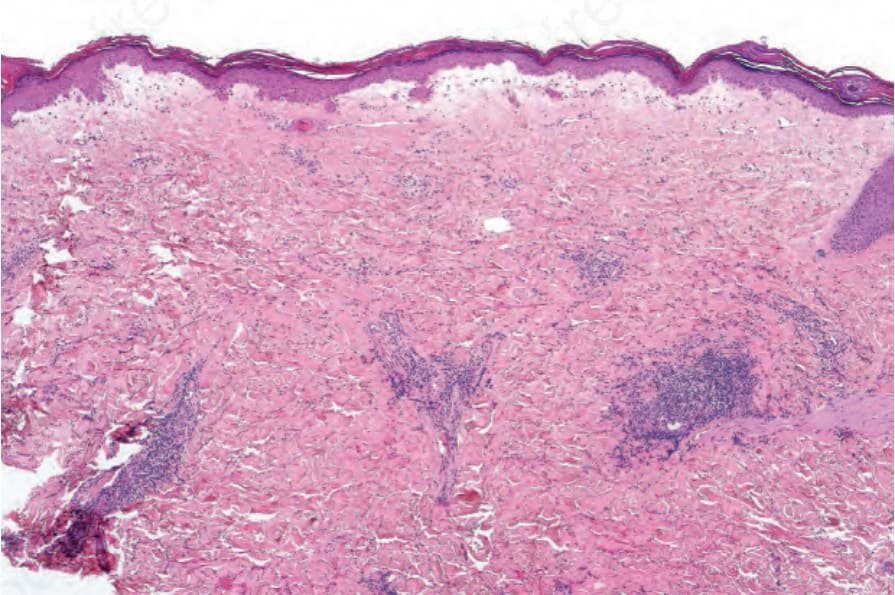
Fig. 17.119 Morphea: intense papillary dermal edema is present, producing a lichen sclerosuslike appearance.
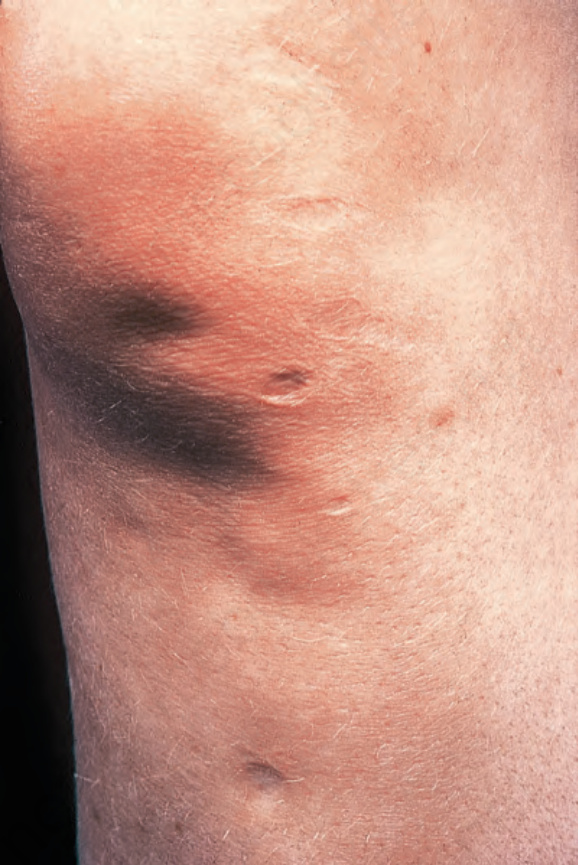
Fig. 17.120 Atrophoderma: lesions present as depressed atrophic plaques with a typical cliffdrop border. By courtesy of the Institute of Dermatology, London, UK.
Phenylketonuria has also been reported to show sclerodermatous features.2
Histologic distinction between morphea and late lesions of acrodermatitis enteropathica may be difficult and occasionally impossible.183 Close clinicopathological correlation allows distinction between these entities.