Eosinophilic granulomatosis with polyangiitis — Part 2
731 Microscopic polyangiitis
• The neutrophil dominates the inflammatory cell infiltrate in polyarteritis nodosa, whereas in eosinophilic granulomatosis with polyangiitis is the eosinophil.
• Necrotizing extravascular granulomata are not a feature of polyarteritis nodosa.
• Patients with granulomatosis with polyangiitis present with ulceroproliferative lesions of the upper respiratory tract, chest pain, and hemoptysis rather than asthma.
• Marked eosinophilia is uncommon in granulomatosis with polyangiitis. Churg-Strauss granulomata may be seen in granulomatosis with polyangiitis; however, the necrosis is more often of the tuberculocoagulative type. Granulomatous vasculitis is not a feature of eosinophilic granulomatosis with polyangiitis, though is not a sensitive feature for discrimination.
can prove fatal.7,8 In one large series from a single institution, over 80% of patients had pulmonary symptoms, suggesting pulmonary involvement may be more common than previously recognized.9
Cutaneous involvement is seen in in approximately 40% of patients.2–6 Dermatological signs include purpura, erythema, splinter hemorrhages, and leg ulceration.6 Bullous presentation and urticaria are occasionally encountered but cutaneous nodules and livedo are rare features of this disease due to absence of involvement of larger vessels.10–12 Other manifestations such as nervous system lesions, gastrointestinal bleeding with pain, and diarrhea, are sometimes evident.13–16
Microscopic polyangiitis is a member of the ANCA-associated vasculitides. Microscopic polyangiitis is usually associated with positive neutrophil cytoplasmic antibodies, typically of the antimyeloperoxidase (perinuclear-antineutrophil cytoplasmic antibody, p-ANCA) subtype (Fig. 16.44).17 In microscopic polyangiitis, about 70% of patients have ANCA directed against MPO (MPO-ANCA), while the remainder have proteinase 3 (PR3)-ANCA.4
It must be stressed that Churg-Straus granulomata should not be taken as pathognomonic for eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). Churg-Strauss granulomata, or nearly identical lesions, has been described in the setting of other systemic diseases including rheumatoid arthritis, lupus erythematosus, other forms of vasculitis (granulomatosis with polyangiitis, polyarteritis nodosa, Takayasu arteritis), lymphoproliferative disorders, Crohn disease and ulcerative colitis, bacterial endocarditis, and hepatitis.46,70–74
Other laboratory findings in microscopic polyarteritis include a raised ESR, normochromic normocytic anemia, leukocytosis with neutrophilia and thrombocytosis, raised C-reactive protein, and raised α-1 and α-2 globulins. Rheumatoid factor and immune complexes are present in less than 50% of patients.5 Anti-DNA antibodies are not a feature. Cutaneous immunofluorescence is usually negative.
This disease is of particular importance due to its high morbidity and mortality, with a 5-year survival of approximately only 65%.5 As a result of improved medical treatment outcome has significantly improved over the past few decades, with a quoted 5-year survival of 81%.18 Severe renal disease and disease relapse are the best indicators for poor prognosis.18
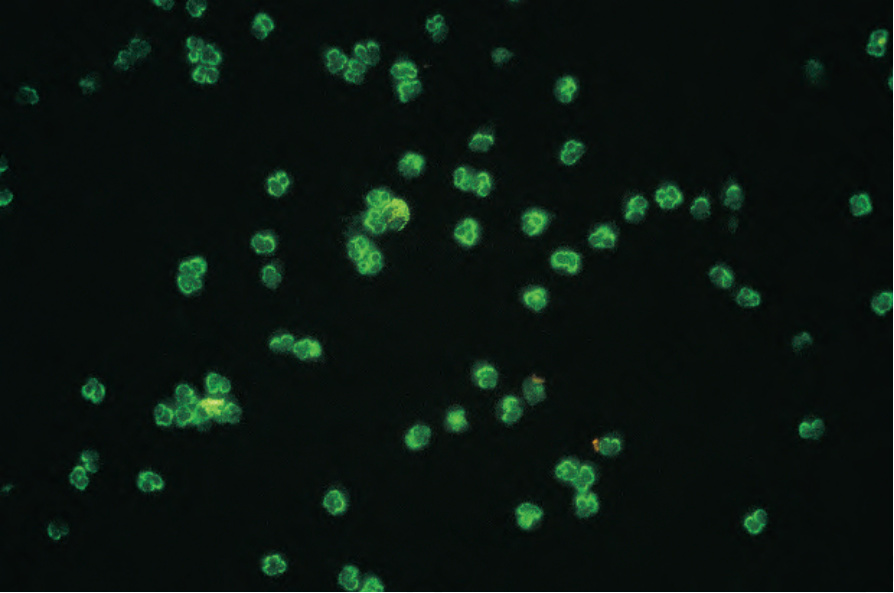
Fig. 16.44 Microscopic polyarteritis: p-ANCA. By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.