Eosinophilic granulomatosis with polyangiitis
Eosinophilic granulomatosis with polyangiitis
Clinical features Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) is a very rare disease that combines the features of asthma, fever, multisystem necrotizing vasculitis, extravascular granulomata, and hypereosinophilia.1,2 Although there is clinical overlap, it can be distinguished from polyarteritis nodosa and granulomatosis with polyangiitis (see Table 16.4). The criteria published by the Chapel Hill Consensus conference differ somewhat.3 In this scheme, eosinophilic granulomatosis with polyangiitis is defined as ‘eosinophil-rich and granulomatous inflammation involving the respiratory tract, and necrotizing vasculitis affecting small to medium-sized arteries, and associated with asthma and eosinophilia’.3 Given differences in classification criteria, it comes as no surprise that inconsistencies between these classification schemes exist.4,5 One study found good concordance between classification schemes for the diagnosis of granulomatosis with polyangiitis
but not eosinophilic granulomatosis with polyangiitis.4 As we gain further understanding of this disease, refinement of diagnostic criteria is likely.
Eosinophilic granulomatosis with polyangiitis may present in a wide range of age groups, but most patients are adults, those in the third and fourth decades being most commonly affected. The disease has a slight male predominance.6–9 Presentation in children is very rare, and most commonly involves teenaged patients.10–14
Asthma and necrotizing vasculitis are almost invariably present. Asthma often precedes the onset of vasculitis, sometimes by many years, or these features develop simultaneously. In one large study, asthma preceded definitive diagnosis in 94% of patients.1 Asthma may be associated with transient pulmonary infiltrates (Loeffler syndrome) or there can be full-blown chronic eosinophilic pneumonitis.6 There is some evidence to suggest that patients in whom vasculitis occurs rapidly after presentation of asthma have a particularly poor prognosis. It has been suggested that, in some cases, treatment for allergic rhinitis with steroids suppresses the full-blown syndrome.15,16 An association between treatment of asthma with antileukotrienes and development of eosinophilic granulomatosis with polyangiitis has been suggested.2,17 The association is controversial and what role, if any, antileukotrienes play in development of disease in these patients is unclear. However, it has also been proposed that it is the withdrawal of the steroids and not the administration of antileukotrienes that leads to disease.2 Further investigation is required to resolve this controversy. Eosinophilic granulomatosis with polyangiitis has also been described following treatment with anti-IgE antibodies (omalizumab) for asthma and may represent unmasking of the disease while reducing steroid treatment.18,19
729 Eosinophilic granulomatosis with polyangiitis
Common manifestations of upper respiratory tract involvement include allergic rhinitis (which is sometimes associated with polyp formation and sinusitis) and hay fever. A family history of atopy and allergic reactions to inhaled antigens and drugs is often present.
Chest radiography frequently confirms the presence of pulmonary involvement, which takes a variety of forms including transient patchy infiltrates, discrete noncavitating nodular masses, or diffuse interstitial disease. On CT scan, pulmonary infiltrates may take the form of opacification, nodules, or bronchial wall and interlobular septal thickening.20 Bronchoalveolar lavage reveals alveolar eosinophilia.1 Certain patients develop an eosinophil-rich pleural effusion.1
ulceration.42–48 Cutaneous infarction and bullae are less common manifestations.46,49 Livedo reticularis involving the lower limbs is occasionally a feature. Patients may also develop tender nodules, which particularly affect the extensor aspects of the arms, legs, hands, and feet (Fig. 16.38). The sacrum, buttocks, and scalp can also be involved. The cutaneous lesions tend to appear in crops with spontaneous relapses and remissions.
Eosinophilic granulomatosis with polyangiitis has been seen in association with HIV infection, hepatitis B, Wells syndrome, and bronchopulmonary candidiasis.50–53 The disease has also been described in association with drugs including fluticasone and cocaine.54,55
Laboratory investigation usually reveals leukocytosis and a raised ESR in association with peripheral blood eosinophilia.46 Blood eosinophilia often decreases with treatment but some authors stress that such a response should not be taken as evidence that disease activity is under control.56 ANCAs are demonstrated in many patients (see below).
In addition to pulmonary lesions, systemic involvement most commonly affects the heart, nervous system, gut, and kidneys.6 Cardiac lesions may be a cause of dysrhythmia or sudden death. Cardiac manifestations also include valvulopathy, ventricular insufficiency, global cardiac insufficiency, and endomyocarditis.1,21–27 Pericardial effusion was seen in 23% of patients in one study.1 Complications relating to cardiac involvement are the most common cause of death in patients with eosinophilic granulomatosis with polyangiitis. Nearly 40% of deaths are due to cardiac involvement.1
Neurological manifestations are frequent, particularly mononeuritis multiplex and symmetric polyneuropathy.28–31 In one large study, 72% of patients developed mononeuritis multiplex.1,30 Intracerebral hemorrhage or infarction sometimes develops.27,28 Ischemic optic and bilateral trigeminal neuropathy are rare complications.29 Myalgia, epilepsy, hydrocephalus, chorea, and vertigo are further occasional features.31
Pathogenesis and histologic features The etiology and pathogenesis of eosinophilic granulomatosis with polyangiitis is poorly understood. The presence of perinuclear-antineutrophil cytoplasmic antibodies (p-ANCA) in many patients is of considerable interest.57–60 ANCAs are detected in approximately 40% of patients.61–63 The ANCAs seen in these patients usually target MPO.63 However, the various types of ANCA are non-specific, being present in a spectrum of disease.64 Their presence is associated with higher risk of developing glomerulonephritis, peripheral neuropathy, and alveolar hemorrhage while absence of ANCAs is linked to heart disease and fever.62,63,65,66 ANCAs may activate neutrophils, causing degranulation and vascular injury.67 T lymphocytes can also be stimulated, leading to endothelial cell injury.67 As with other ANCA-associated vasculitides, it is suspected that they play a role in the pathogenesis; however, the precise mechanism, particularly triggering factors, is not yet known. Persistence of ANCAs with therapy may be of limited value in making treatment decisions.68 One group has found that, although there is poor correlation between ANCA titer and disease activity, disappearance of ANCA can reflect absent disease activity.69
Evidence of gastrointestinal involvement, such as nausea, bleeding, vomiting, and abdominal pain, is often found. In one study, one-third of patients experienced gastrointestinal symptoms, usually abdominal pain.1 Diffuse bowel ischemia is an uncommon but serious complication.1,33
Renal disease in eosinophilic granulomatosis with polyangiitis is usually manifest as glomerulonephritis, most often a focal segmental glomerulonephritis.1,32 Patients with renal involvement show hematuria, proteinuria, and increased creatinine.1 Renal infarction appears to be a rare complication.1
Rheumatological involvement in the form of polyarthralgia and constitutional symptoms, including fever, anemia, and weight loss, is common.1
Amyloidosis is a rare complication.34–36 Exceptionally, eosinophilic granulomatosis with polyangiitis may present with temporal nongiant cell arteritis.37 Involvement of the breast occurs exceptionally as eosinophilic mastitis.38–40 A limited form of the disease has been described.41,42
Cutaneous lesions are seen in 40% to 70% of patients and include petechiae, purpura, papules, vesicles, facial erythema, urticaria, and
Pulmonary lesions comprise variably sized (up to 1.5 cm) nodules, ranging from only a few lesions to hundreds which may coalesce. Histologically, they are composed of granulomata with central necrosis and surrounding epithelioid histiocytes with occasional giant cells. Large numbers of eosinophils with an admixture of lymphocytes, neutrophils, plasma cells, and histiocytes infiltrate the adjacent lung parenchyma. Vasculitis involving small arteries and sometimes veins is also present.
Cutaneous lesions are variable. A common feature is the so-called Churg-Strauss (extravascular) granuloma. Early lesions are characterized by focal collagen degeneration in association with a varying and mixed
730 Vascular diseases
study, 16 of 37 biopsies (taken from 29 patients) showed leukocytoclastic vasculitis.45 Occasionally, the arteries in the dermis and subcutaneous fat show changes similar to those seen in polyarteritis nodosa.46 Additionally, acute and chronic panniculitis with eosinophils has been described.46
inflammatory cell infiltrate comprising neutrophils, lymphocytes, and histiocytes (Figs 16.39 and 16.40). Eosinophils may be sparse or numerous. Leukocytoclasis is often a feature. In more advanced examples the granuloma is more mature in appearance, consisting of a central zone of collagen necrosis surrounded by a peripheral palisade of epithelioid and giant cells (Figs 16.41 and 16.42). In some examples, the features are those of a rather diffuse and ill-defined granulomatous inflammatory process without obvious collagen degeneration. Commonly, features of necrotizing vasculitis are evident: fibrinoid necrosis accompanied by an eosinophilic and neutrophilic infiltrate with leukocytoclasis involving the more superficial small blood vessels (Fig. 16.43). There may be epidermal ischemic necrosis. In one
Differential diagnosis The histologic features encountered in skin biopsies of patients with eosinophilic granulomatosis with polyangiitis are not diagnostic. Careful clinicopathological and serological evaluation is necessary to establish a definitive diagnosis. Although eosinophilic granulomatosis with polyangiitis, polyarteritis nodosa, and granulomatosis with polyangiitis show both clinical and histologic overlap, research over the last several decades leaves no doubt that they represent distinctive entities. Nonetheless, they form a spectrum of disease with similar pathogenesis, although there are sufficient differences to justify their separate classification:
• Asthma may be seen in both polyarteritis nodosa and eosinophilic granulomatosis with polyangiitis, but characteristically polyarteritis affects medium-sized and small arteries, while eosinophilic granulomatosis with polyangiitis typically affects small arteries and veins.
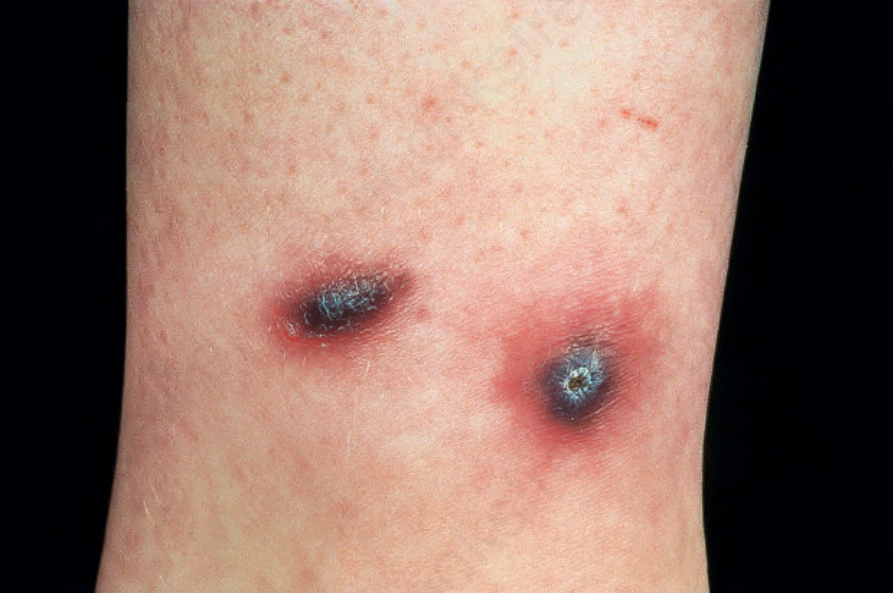
Fig. 16.38 Eosinophilic granulomatosis with polyangiitis: this patient presented with painful nodules on the limbs. By courtesy of the Institute of Dermatology, London, UK.
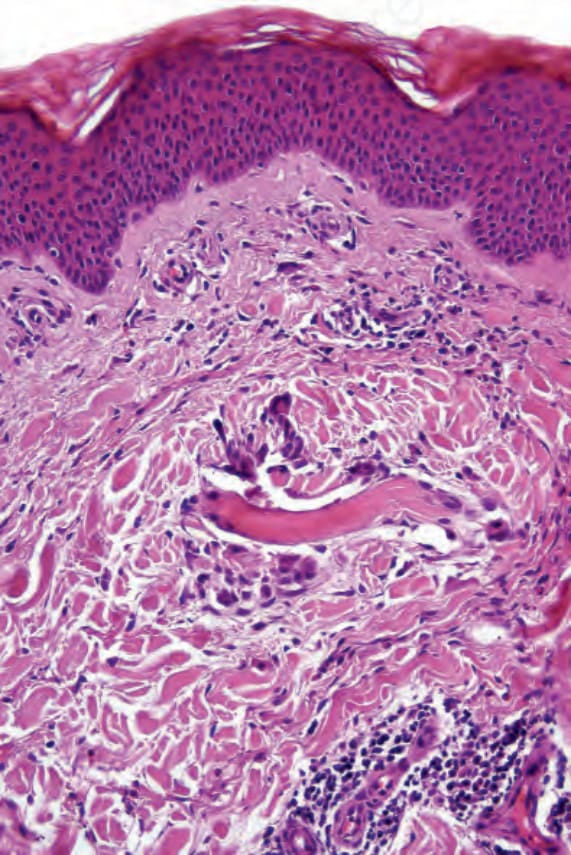
Fig. 16.39 Eosinophilic granulomatosis with polyangiitis: this early lesion shows a swollen collagen fiber in the superficial dermis. Note the surrounding multinucleate giant cells.
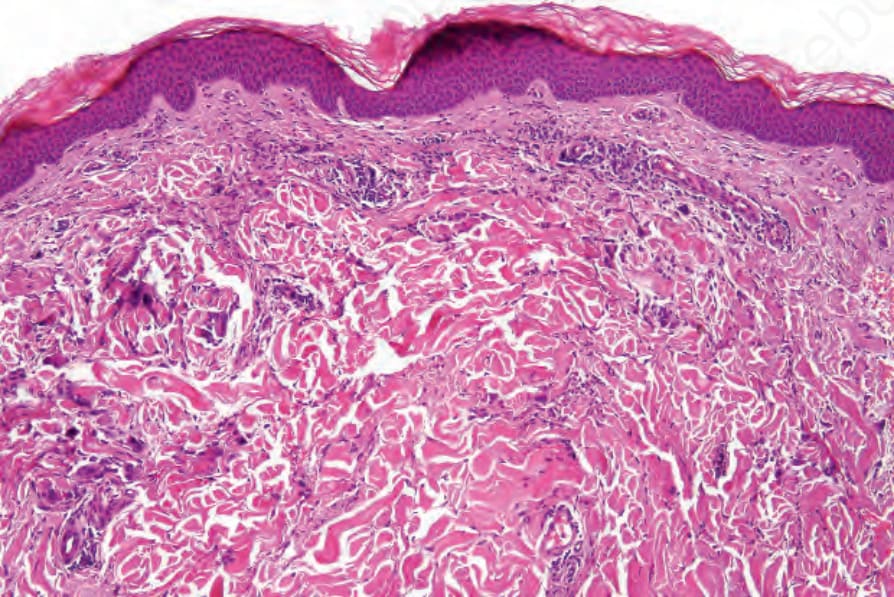
Fig. 16.40 Eosinophilic granulomatosis with polyangiitis: medium-power view showing swelling of the dermal collagen fibers and a perivascular chronic inflammatory cell infiltrate.
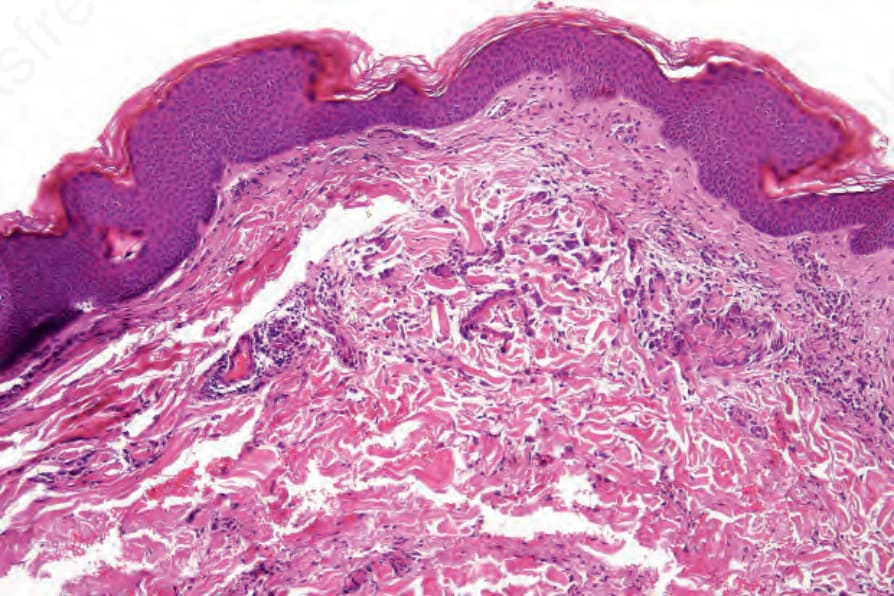
Fig. 16.41 Eosinophilic granulomatosis with polyangiitis: in this field there is a more obvious granulomatous infiltrate.
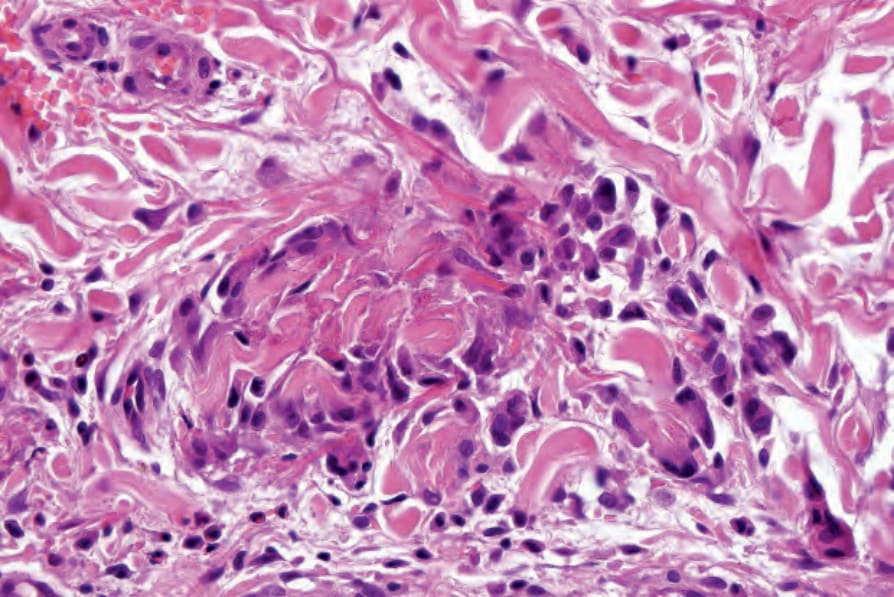
Fig. 16.42 Eosinophilic granulomatosis with polyangiitis: this florid example shows a granulomatous infiltrate containing prominent giant cells. By courtesy of E. Wilson Jones, MD, Institute of Dermatology, London, UK.
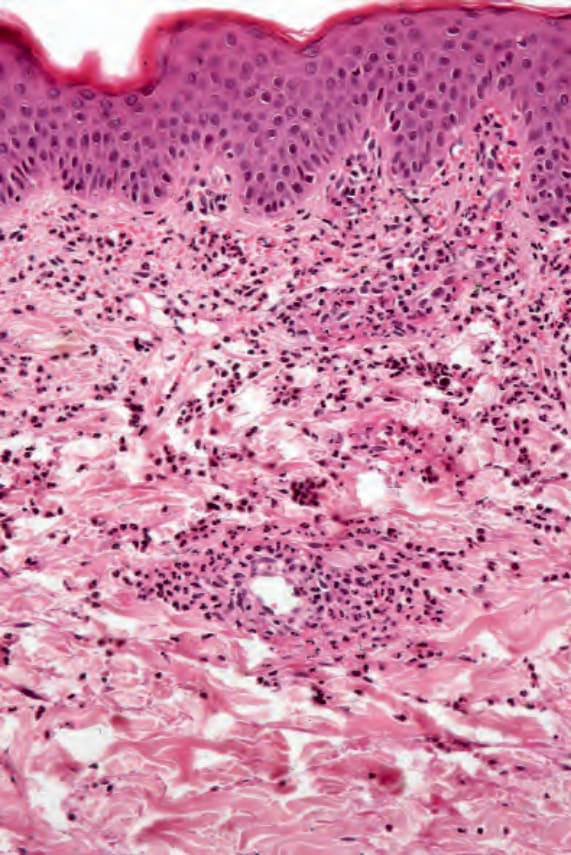
Fig. 16.43 Eosinophilic granulomatosis with polyangiitis: the features of small-vessel leukocytoclastic vasculitis are evident.
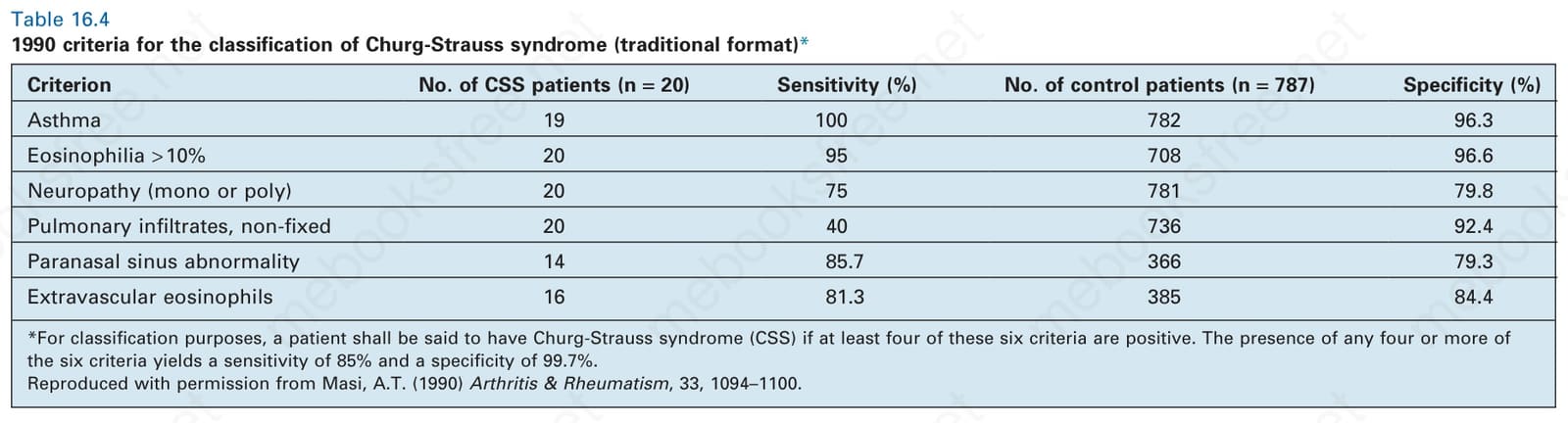
Table 16.4 1990 criteria for the classification of Churg-Strauss syndrome (traditional format)*