Granulomatosis with polyangiitis
Granulomatosis with polyangiitis
Clinical features Granulomatosis with polyangiitis is a multisystem vascular disease associated with high morbidity and mortality.1–3 Before the introduction of
Pulmonary lesions are present in up to 90% and patients may have cough, chest pain, or hemoptysis.12 Radiological examination frequently reveals solitary or more commonly multiple nodular opacities, which are often bilateral, may be diffuse or sharply delineated, and are typically transient. Cavitation is frequently a feature. Lesions may present as large nodules that are clinically and radiologically suspicious for malignancy.
Renal involvement is usually in the form of focal segmental necrotizing glomerulonephritis.12 Urinalysis typically reveals hematuria (often microscopic), proteinuria, and red cell casts.
725 Granulomatosis with polyangiitis
Criterion Definition
Nasal or oral inflammation
Development of painful or painless oral ulcers or purulent or bloody nasal discharge
Abnormal chest radiograph
Chest radiograph showing the presence of nodules, fixed infiltrates or cavities
Urinary sediment Microhematuria (> 5 red blood cells per high-power field) or red cell casts in urine sediment
Granulomatous inflammation on biopsy
Histologic changes showing granulomatous inflammation within the wall of an artery or in the perivascular or extravascular area (artery or arteriole)
*For purposes of classification, a patient shall be said to have Granulomatosis with polyangiitis if at least two of these four criteria are present. The presence of any two or more criteria yields a sensitivity of 88.2% and a specificity of 92.0% Reproduced with permission from Leavitt, R.Y. (1990) Arthritis & Rheumatism, 33, 1101–1107.
In one large series, 34% of patients developed neurological involvement.13 Peripheral neuropathy was seen in 16%.13 Central nervous system (CNS) lesions are not uncommon and occur either as a consequence of direct extension through the base of the skull from sinus involvement or as a result of meningeal or intracerebral necrotizing granulomata. Patients may experience myelopathy or neuropathy.14 Vasculitis involving intracerebral vessels can also result in cerebral lesions. Patients develop cranioneuropathy, cerebrovascular accidents, or seizures.13 Involvement of the vasa nervosa may give rise to mononeuritis multiplex.
In addition to the organ-specific features noted above, patients also often have a variety of constitutional symptoms, including anorexia, weight loss, fever, and general malaise.
Two limited forms are recognized: pathergic granulomatosis and limited pulmonary granulomatosis.22–24
• Pathergic granulomatosis is of particular importance because mucosal and cutaneous lesions may predominate and persist for very long periods of time before intractable renal failure develops. In the absence of evidence of pulmonary and renal involvement, there may be a delay in establishing the diagnosis and administration of appropriate chemotherapy, with resultant increased morbidity and mortality. Patients with this variant are at particular risk of facial mutilation; sites especially involved include the nose, nasopharynx, sinuses, and middle ears (Fig. 16.29).
• In limited pulmonary granulomatosis patients have respiratory symptoms with associated fever and weight loss. Radiologically, multiple bilateral discrete nodular infiltrates and thin-walled cavitating lesions are seen, usually in the lower lobes. No evidence for renal involvement is present. Patients with this variant appear to have a somewhat better prognosis than those with classic (generalized) granulomatosis with polyangiitis.
Ocular lesions result in a variety of complications including conjunctivitis, granulomatous keratitis, sclerouveitis, and orbital pseudotumor. Proptosis is sometimes a feature.15 Involvement of the temporal artery results in features (i.e., vision loss, jaw claudication) similar to those seen in temporal arteritis.16
Cutaneous manifestations are common, occurring in about 10% to 50% of patients.12,17–20 Several different types of skin lesion may be encountered, including vasculitic lesions with purpura, bruising, and nodule formation (Figs 16.26–16.28). Pyoderma gangrenosum-like lesions with necrosis and ulceration that have a predilection for the lower limbs are sometimes encountered. The presence of skin lesions appears to correlate with disease activity. Oral ulceration is common.20,21
726 Vascular diseases
arteritis, eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), Kawasaki arteritis, microscopic polyangiitis, and idiopathic crescentic glomerulonephritis.28,32,33
Pathogenesis and histologic features This rare disease is thought to represent a hypersensitivity reaction triggered by a variety of stimuli, including infections, environmental factors, and medications in genetically susceptible patients.34. Response to immunosuppressive therapy and the increased risk for patients with MHC class II HLA-DP1*0401, MHC class II HLA-DRB1 ∗ 15, or MHC class II HLADRB ∗1501 are consistent with this hypothesis.35 The presence of ANCAs against PR3 and to a lesser amount MPO in most patients with granulomatosis with polyangiitis and the correlation of circulating levels of ANCAs with disease activity supports a role in the pathogenesis of this disease, as do animal models of ANCA-associated vasculitis.36 Additionally, although immune complexes have not been demonstrated, disease activity is ameliorated with plasma exchange. Thus, there is compelling evidence suggesting ANCAs are central to pathogenesis, most likely through activation of neutrophils, lymphocytes, and macrophages.37 In particular, abnormal numbers and function in regulatory T cells (Treg) have been demonstrated in patients with granulomatosis and polyangiitis and appear to also correlate with disease activity.38–40 However, the precise mechanism action of ANCAs is not yet fully understood.37
It is postulated that exposure to an antigen (or antigens) may trigger ANCAs that have pathophysiological effects leading to tissue destruction.34,41 Infectious agents have received some attention as potentially playing a role in the pathogenesis of granulomatosis with polyangiitis. It is interesting to note that relapses of the disease may follow infection.37 In some patients, a complete or partial remission is achieved with antibiotic treatment combined with immunosuppressive agents.37,42 Trimethoprim-sulfamethoxazole has also been used to reduce the frequency of relapses in patients with granulomatosis and polyangiitis.43 Patients who are chronic nasal carriers of S. aureus seem to have a higher relapse rate compared with noncarriers.44 Furthermore, antibodies against hepatitis C virus, Epstein-Barr virus, and H. pylori as well as IgG antibodies against Toxoplasma gondii and IgM antibodies against cytomegalovirus are significantly more common in patients with granulomatosis with polyangiitis than in unaffected individuals.45 Gastrointestinal and renal manifestation correlates well with the presence of IgG antibodies to cytomegalovirus while otolaryngeal manifestation is more common in patients with IgG antibodies to the Epstein-Barr virus early antigen.45 Despite considerable research to establish a possible relationship between granulomatosis with polyangiitis and infection, a categoric role in the disease is elusive.46
It has been suggested that the presence of extravascular granulomata (particularly affecting the ears, nose, throat, orbit, or lung) in association with a positive serum cytoplasmic-antineutrophil cytoplasmic antibody (c-ANCA) represents the earliest stage in the evolution of granulomatosis with polyangiitis.25 There are cases in which patients had relatively minor symptoms in which serological testing for c-ANCA helped establish the diagnosis.26 There are also case reports of patients with smoldering symptoms that fully develop vasculitis after many years.27 While these cases are exceptional, it does suggest that some cases may be diagnosed at an early stage before the development of more serious multisystem disease and, hence, earlier treatment.
In addition to that for infectious agents, a search for putative roles for physical agents in the environment has also been undertaken. Perhaps most attention has focused on silicon compounds.47–49 One case-control study showed that exposure to silicon-containing compounds conferred a sevenfold risk for the development of granulomatosis with polyangiitis.48 It has been postulated that silica-induced apoptosis of inflammatory cells may release lysosomal enzymes that stimulate ANCAs.37,47
Pulmonary lesions are characterized by necrotizing granulomatous inflammation that may bear more than a superficial resemblance to the caseation of pulmonary tuberculosis (Fig. 16.31).50 The similarity is increased by the presence of large numbers of Langhans giant cells at the periphery of the necrotic focus (Fig. 16.32). In addition, the features of an active angiitis are present; this may involve both arteries and veins and frequently has a granulomatous component (Fig. 16.33). The adjacent parenchyma is chronically inflamed and often shows severe, diffuse, interstitial fibrosis.
Greater than 90% of patients with granulomatosis and polyangiitis have ANCA detected in their sera; rising titers have been shown to correlate with disease activity and are a valuable method of predicting relapse.2,28,29 Typically, the indirect immunofluorescence shows a cytoplasmic pattern of staining (c-ANCA) (Fig. 16.30).1 In the majority (70–80%) of patients with active disease ANCAs are directed against proteinase 3 (PR3) while ANCAs against myeloperoxidase (MPO) are detected in approximately 10% of patients.30,31 Some patients who are negative for ANCAs by conventional methods have antibodies to lysosome-associated membrane glycoprotein 2, while others have antibodies for a MPO peptide not detected by conventional methods.29 ANCAs have also been detected in patients with Takayasu
Early renal lesions are characterized by focal segmental glomerulonephritis. In more advanced cases the glomerulitis becomes generalized, with fibrinoid necrosis and widespread epithelial crescent formation.51 The renal interstitial tissue may contain necrotizing granulomata, and vasculitis is sometimes a feature. Immunofluorescence occasionally reveals granular deposits of immunoglobulin and complement along the glomerular capillary walls. This is taken as evidence for possible immune complex involvement. Similar granulomata and evidence of vasculitis have been described in all organ systems of the body, but are particularly often seen in the spleen.
727 Granulomatosis with polyangiitis
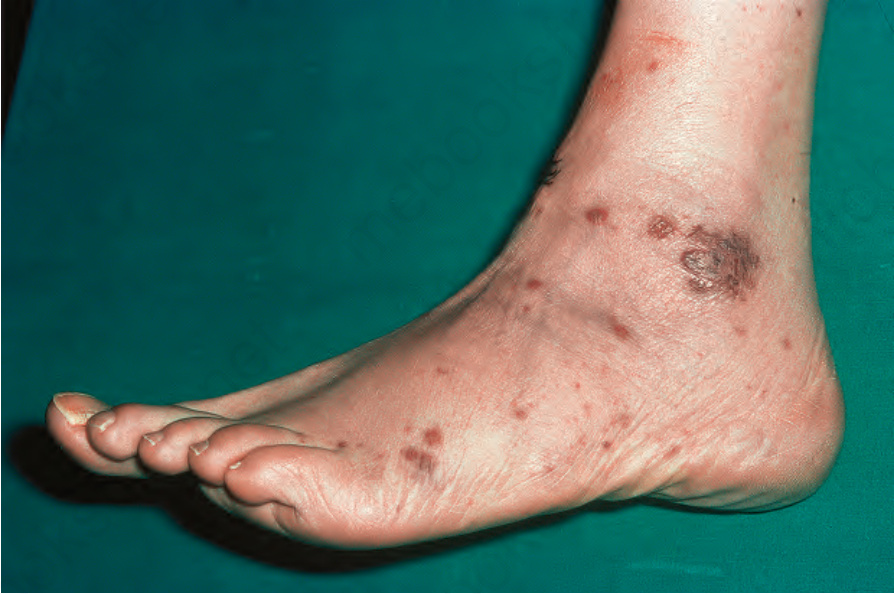
Fig. 16.26 Granulomatosis with polyangiitis: multiple purpuric macules and papules. By courtesy of D. McGibbon, MD, St Thomas’ Hospital, London, UK.
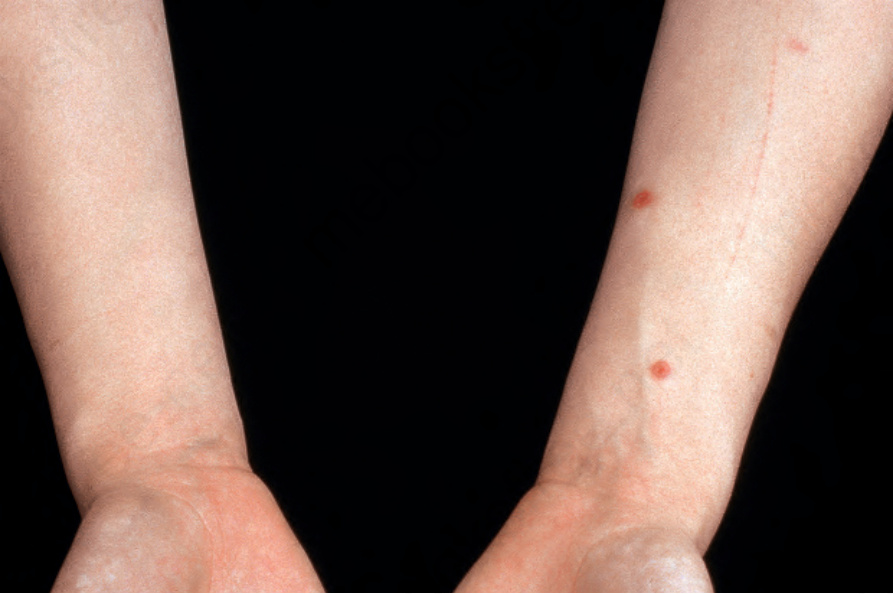
Fig. 16.27 Granulomatosis with polyangiitis: cutaneous nodules as seen in this patient are a not uncommon manifestation. By courtesy of the Institute of Dermatology, London, UK.
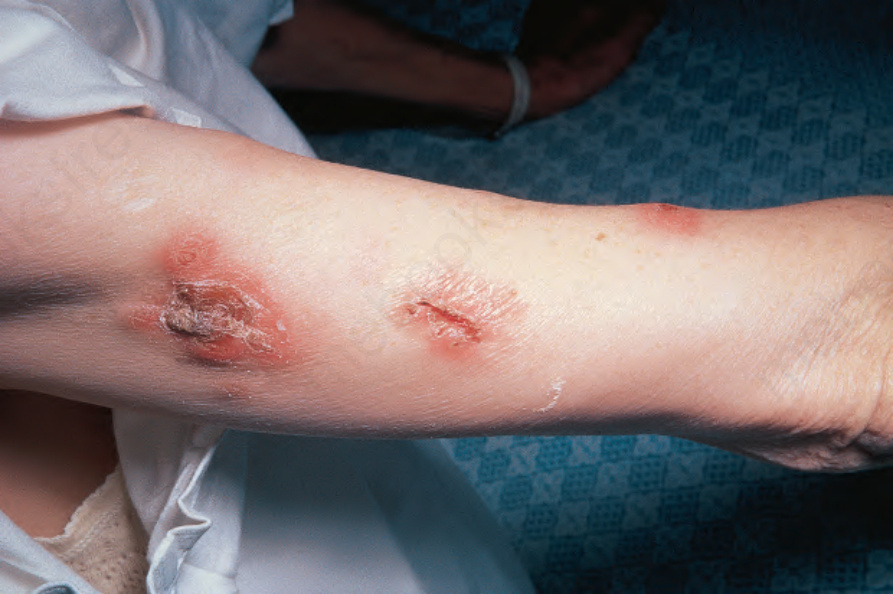
Fig. 16.28 Granulomatosis with polyangiitis: this patient has ulcerating plaques and nodules. By courtesy of the Institute of Dermatology, London, UK. Joint involvement may present as arthralgia or, less commonly, frank arthritis.
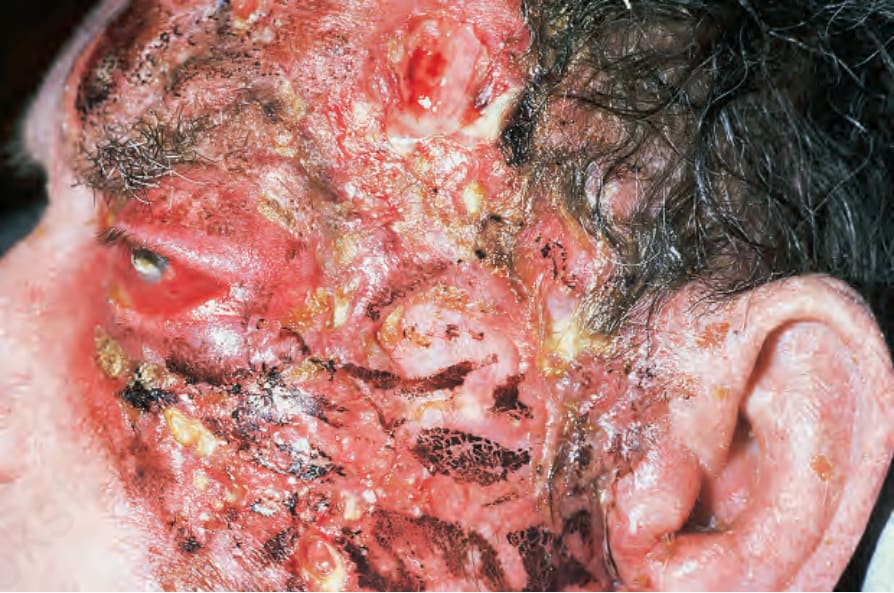
Fig. 16.29 Pathergic granulomatosis: gross necrosis and ulceration have resulted in very disfiguring tissue damage.
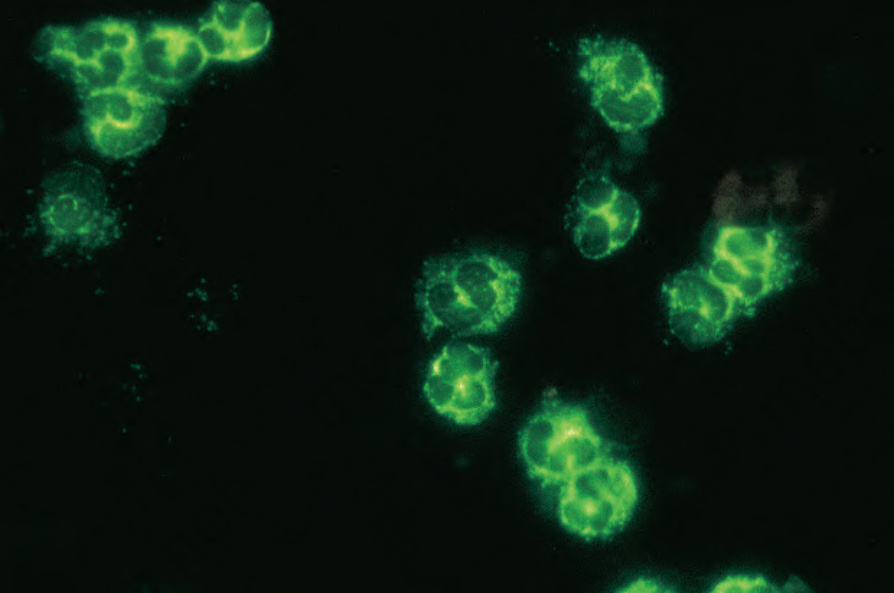
Fig. 16.30 Granulomatosis with polyangiitis: c-ANCA. By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
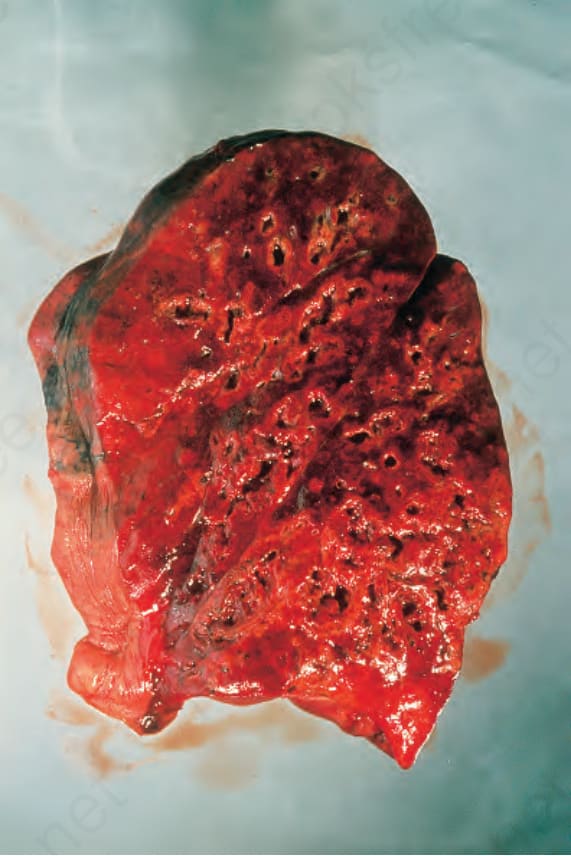
Fig. 16.31 Granulomatosis with polyangiitis: this postmortem lung specimen shows consolidation and numerous abscesses. By courtesy of B. Corrin, MD, Brompton Hospital, London, UK.
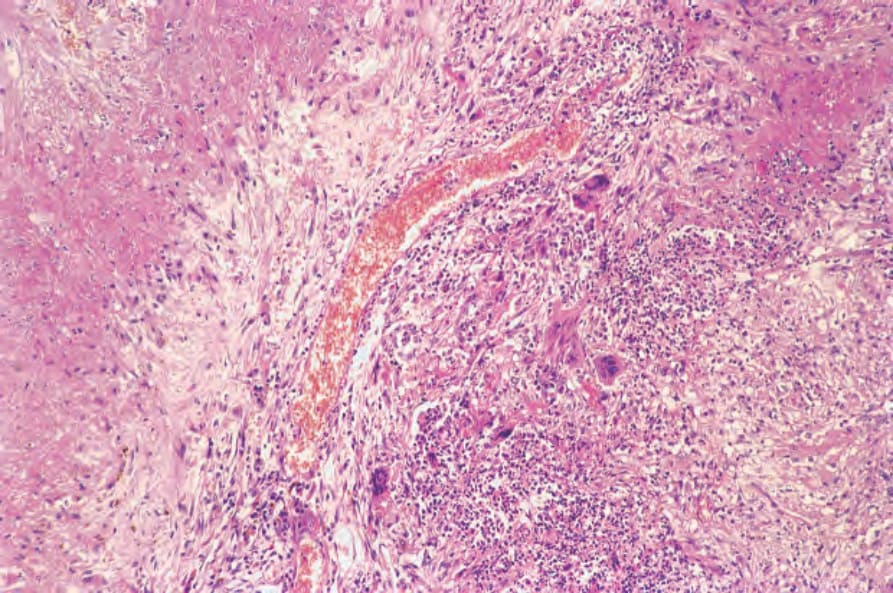
Fig. 16.32 Granulomatosis with polyangiitis: this lung section shows extensive necrosis associated with a granulomatous infiltrate containing Langhans giant cells. These appearances resemble pulmonary tuberculosis.
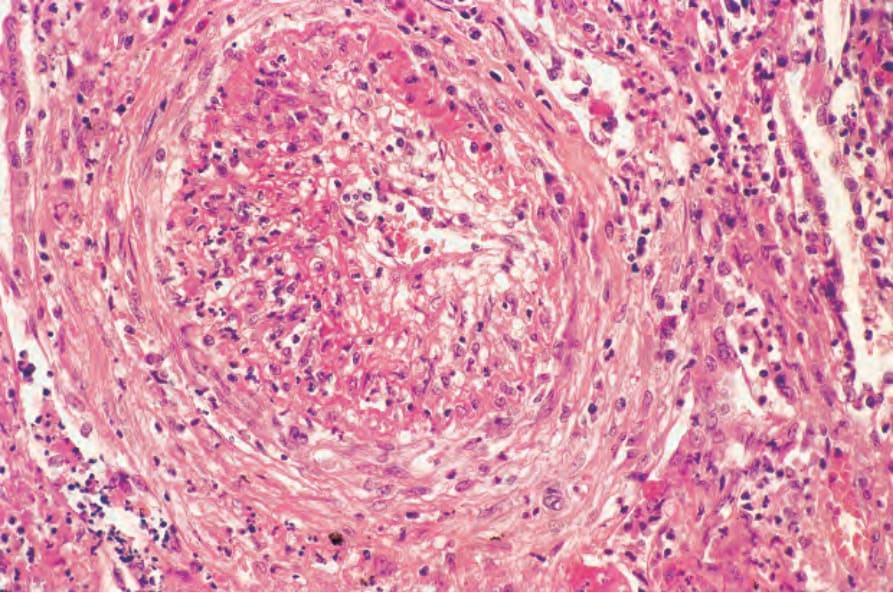
Fig. 16.33 Granulomatosis with polyangiitis: a branch of the pulmonary artery shows necrotizing arteritis with fibrointimal thickening.
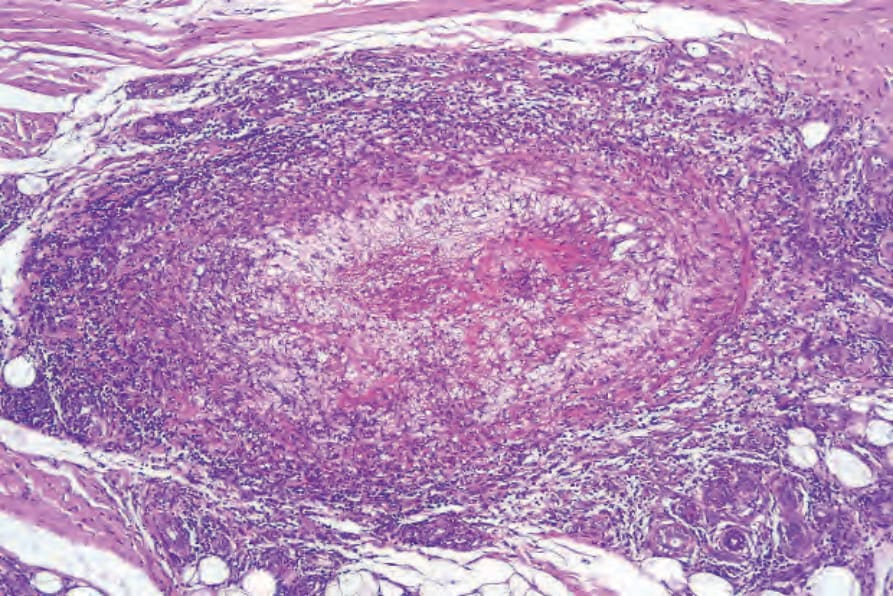
Fig. 16.35 Granulomatosis with polyangiitis: large vessel showing intense chronic inflammation, thrombosis, and intimal fibrosis.
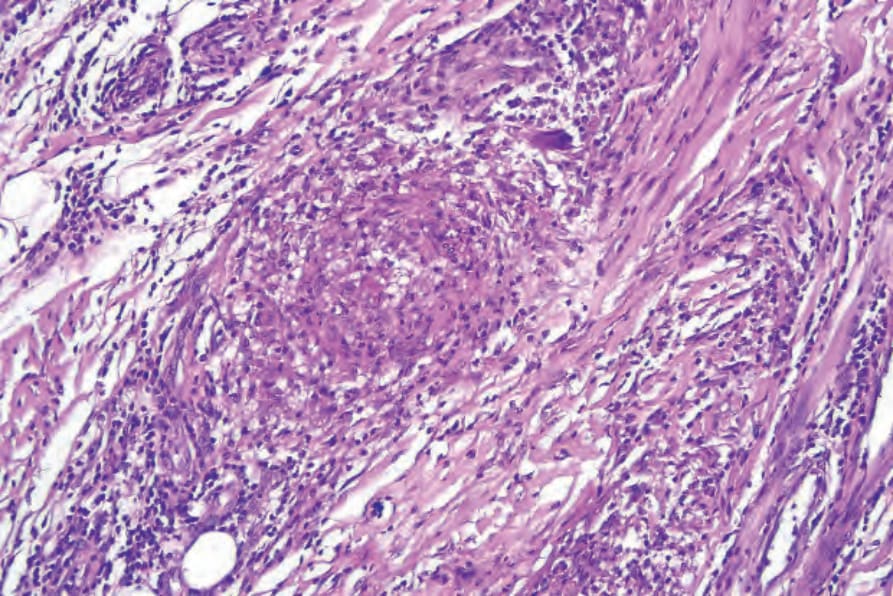
Fig. 16.37 Granulomatosis with polyangiitis: high-power view of an ill-defined granuloma.
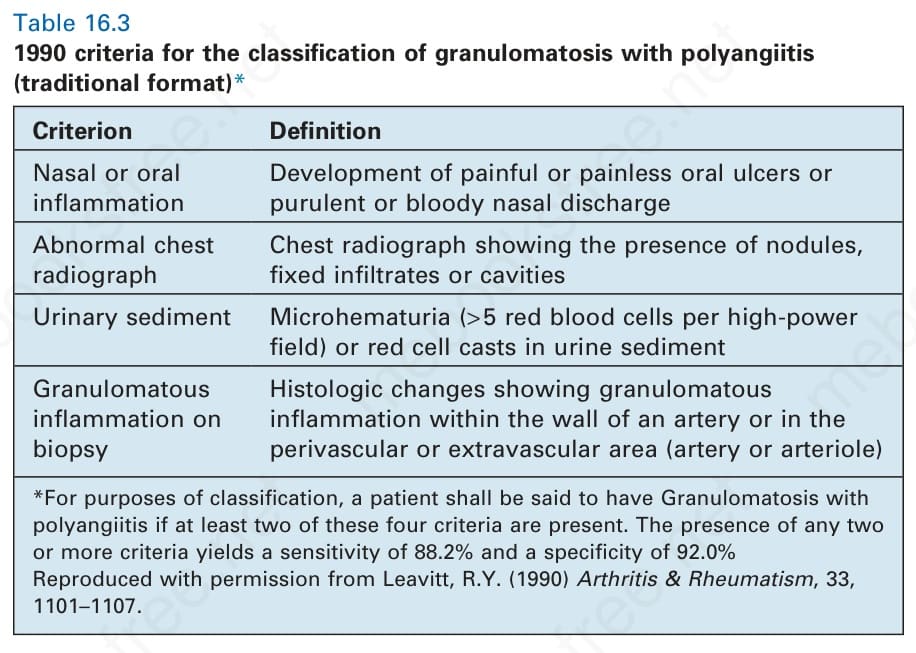
Table 16.3 1990 criteria for the classification of granulomatosis with polyangiitis (traditional format)*
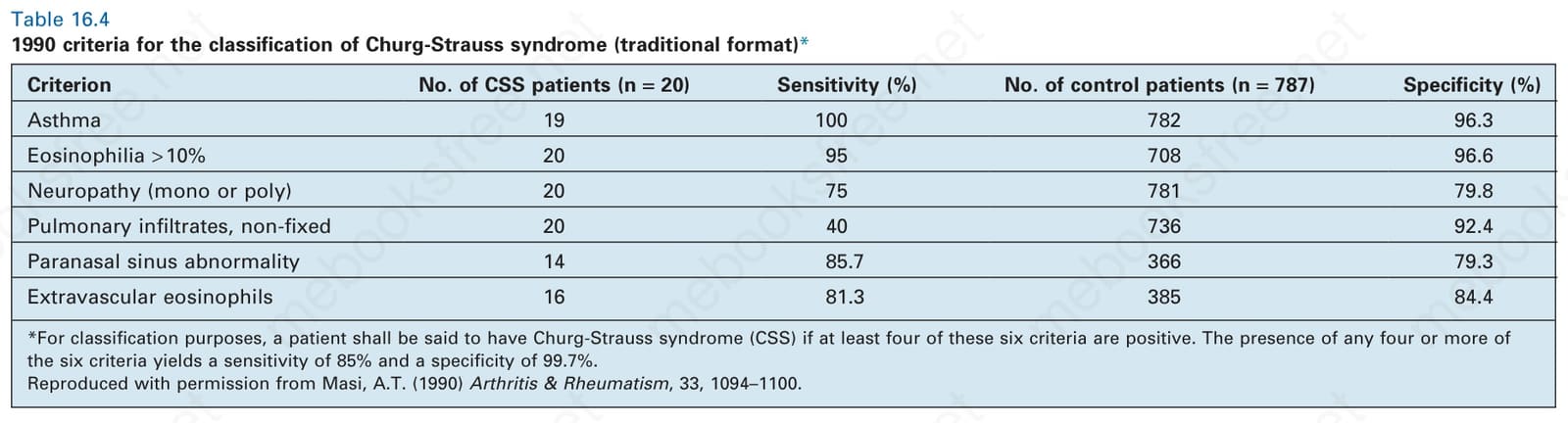
Table 16.4 1990 criteria for the classification of Churg-Strauss syndrome (traditional format)*
Cutaneous lesions reveal a variety of features, including necrotizing vasculitis, in which small and/or medium-sized dermal vessels display fibrinoid necrosis, a neutrophil polymorphonuclear infiltrate, and leukocytoclasis (Figs 16.34 and 16.35). In one series, 80% of biopsies from patients with cutaneous lesions (of 244 patients in this series, 14% had cutaneous lesions) showed leukocytoclastic vasculitis.19 In another study, nearly a third showed leukocytoclastic vasculitis and another third showed non-specific chronic inflammation.52 In this study, nearly 50% of patients had entirely non-specific findings. Extravasated red blood cells are invariably present.17,18 In severe cases, the epidermis may show ischemic necrosis. Bone fide granulomatous vasculitis of skin appears to be a very rare feature.52,53 In fact,
728 Vascular diseases
Criterion No. of CSS patients (n = 20) Sensitivity (%) No. of control patients (n = 787) Specificity (%)
Asthma 19 100 782 96.3
Eosinophilia > 10% 20 95 708 96.6
Neuropathy (mono or poly) 20 75 781 79.8
Pulmonary infiltrates, non-fixed 20 40 736 92.4
Paranasal sinus abnormality 14 85.7 366 79.3
Extravascular eosinophils 16 81.3 385 84.4
*For classification purposes, a patient shall be said to have Churg-Strauss syndrome (CSS) if at least four of these six criteria are positive. The presence of any four or more of the six criteria yields a sensitivity of 85% and a specificity of 99.7%. Reproduced with permission from Masi, A.T. (1990) Arthritis & Rheumatism, 33, 1094–1100.
one study failed to demonstrate granulomatous vasculitis in 75 skin biopsies from 46 patients.52 In other patients, there may be granulomatous infiltration of the dermis, which may be related to foci of collagen necrosis and sometimes resembles the granulomas seen in eosinophilic granulomatosis with polyangiitis (Figs 16.36 and 16.37). In some cases, extensive geographic zones of necrosis are present, associated with a mixed inflammatory cell infiltrate including variable numbers of histiocytes, giant cells, lymphocytes, eosinophils, and plasma cells. Erythema nodosum and granuloma annulare-like lesions may also be encountered.52
Differential diagnosis As mentioned above, early in the course of the disease, when patients may not have developed the full clinical triad, definitive diagnosis is sometimes impossible.
When granulomata and/or allergic vasculitis are the only features, it may not be possible to histologically distinguish granulomatosis with polyangiitis from eosinophilic granulomatosis with polyangiitis. A high eosinophil content, however, is somewhat suggestive of the latter condition but certainly not diagnostic, as this finding may sometimes be seen in granulomatosis with polyangiitis.54 Therefore, distinction of granulomatosis with polyangiitis from other forms of granulomatous inflammation and leukocytoclastic and granulomatous vasculitis requires careful clinicopathological and serological correlation.
In those instances where a granulomatous dermal infiltrate occurs in the absence of vasculitis, a host of conditions enters the differential diagnosis, including sarcoidosis and infections, particularly mycobacterial and fungal. Granulomatous vasculitis may also be seen in association with lymphoproliferative diseases, including lymphoma, angioimmunoblastic lymphadenopathy, and leukemia.54
Microscopic polyangiitis can be confused with granulomatosis with polyangiitis. The presence of granulomatous inflammation in the lung would favor the latter. Microscopic polyangiitis is approached as a diagnosis of exclusion, as mentioned previously. In fact, a diagnosis may be revised as the pattern of clinical involvement changes. For example, patients who appear to fit criteria for microscopic polyangiitis may eventually develop manifestation allowing for classification as granulomatosis with polyangiitis.55
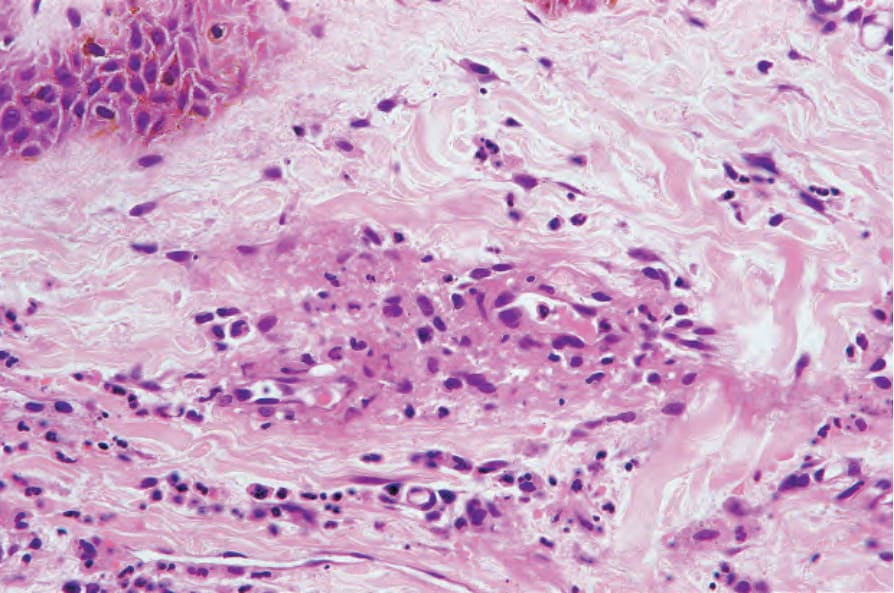
Fig. 16.34 Granulomatosis with polyangiitis: leukocytoclastic vasculitis as shown in this field is the most frequently encountered cutaneous lesion.
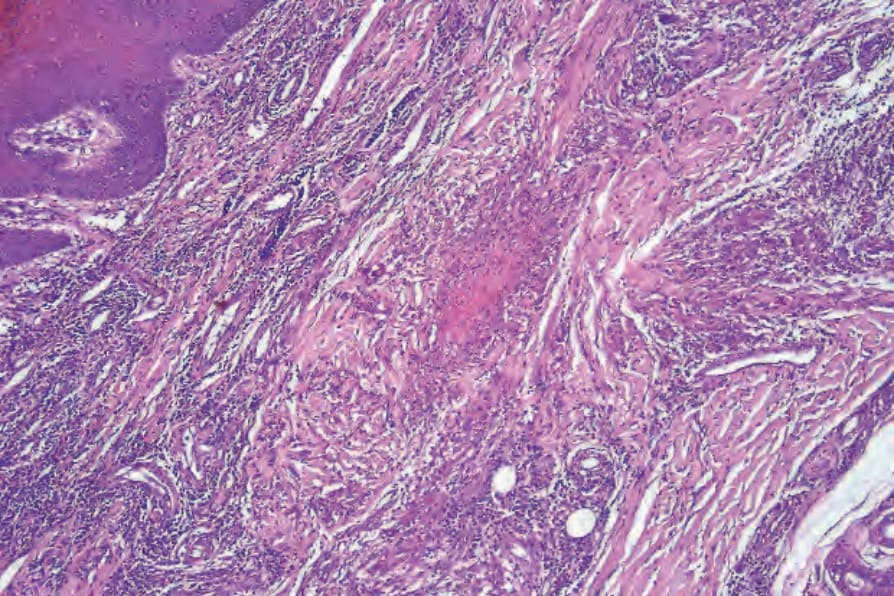
Fig. 16.36 Granulomatosis with polyangiitis: low-power view of a necrotizing dermal granuloma.