IgA vasculitis (Henoch-Schönlein purpura)
IgA vasculitis (Henoch-Schönlein purpura)
Clinical features IgA vasculitis (Henoch-Schönlein purpura) is a syndrome characterized by abdominal pain, joint symptoms, and palpable purpura secondary to leukocytoclastic vasculitis, and caused by circulating IgA immune complexes. The disease typically involves children (males more often than females), although adults may also be affected.1–6 Occurrence during pregnancy has only rarely been documented.7 In a large study of children with Henoch-Schönlein purpura, 92% of patients were less than 10 years of age.8
It often complicates an upper respiratory tract infection and is characterized by a seasonal incidence with a peak in winter.1 Clustering of cases has been described, leading one group of authors to postulate that person-to- person spread of an infectious agent plays a role in the pathogenesis of this syndrome.9 Although it may follow a streptococcal throat infection, it sometimes develops after a wide variety of other infective conditions including amebiasis, chickenpox, hepatitis, HIV, yersiniosis, and infection by Toxocara canis, Helicobacter pylori, Pseudomonas aeruginosa, Staphylococcus aureus, Escherichia coli, and erythrovirus (formerly parvovirus) B19.10–15 In one series > 30% had documentation of some sort of an infection prior to the development of vasculitis.5 Additional causes include adverse reactions to drugs such as ampicillin, penicillin, erythromycin, clarithromycin, glyburide, etanercept, infliximab, and adulmumab.16–22 An association with cocaine inhalation has also been described.23 In one study, drug therapy may have been a precipitating cause in 26% of patients.24
As noted above, classic IgA vasculitis is characterized by a triad of purpura, abdominal pain, and arthralgia. The cutaneous clinical findings are those of leukocytoclastic vasculitis. Cutaneous lesions are most frequently the presenting symptom and comprise palpable purpura predominantly affecting the lower limbs, thighs, and buttocks (Fig. 16.21).1,5,25–27 Targetoid lesions are often present.28 Hemorrhagic bullae are uncommon.5,29–31 Rare pustules and subcutaneous nodules have also been documented.5,32 A prodrome of itchy urticaria is sometimes described.2 Children often have edema, particularly of the feet and lower legs, although it may be more widespread.
In patients with associated hypocomplementemia, neutrophils are predominant with far fewer lymphocytes; patients who are normocomplementemic may show lymphocyte predominance. In the surrounding connective tissue, red cell extravasation, edema, and an inflammatory neutrophil infiltrate associated with karyorrhexis (leukocytoclasis) are typically present (Fig. 16.20).
The severity of the histopathological changes in the cutaneous lesions of leukocytoclastic vasculitis does not predict extracutaneous involvement.139
Differential diagnosis The diagnosis is relatively straightforward. It is critical to understand that leukocytoclastic vasculitis is not a disease sui generis. Rather, it represents a reaction pattern due to circulating immune complexes that may be caused
In one large study, arthritis was seen in 82% of patients and was the presenting feature in 24%.8 Joint involvement consists of migratory arthralgia predominantly affecting the large joints of the lower limbs. Involvement of the upper extremity occurred in 37%, with the hand and wrist being more often affected than the elbow.8
Intestinal involvement with resultant red cell extravasation or hemorrhage leads to abdominal pain and gastrointestinal bleeding. Abdominal pain was noted in 63% of patients in one study of 100 consecutive children presenting with the condition.8 Gastrointestinal disease develops as a consequence of acute vasculitis. Bleeding may be either occult or in the form of bloody stools.8 Intussusception is an occasional complication.33,34 Abdominal pain was the presenting complaint in 19% of patients in one study.8 Endoscopy may reveal hemorrhage, ulceration, and erosions.35 IgA is often noted in capillaries of the gastrointestinal tract but frank necrotizing vasculitis was
722 Vascular diseases
A
B
months.12 In a further series, a third of patients suffered at least one recurrence of symptoms, usually within a few months of initial presentation.8 Recurrences were also found to be more frequent in patients > 8 years of age and in those with nephritis.25
Solid tumors including lung, prostate, breast and gastrointestinal cancer, and hematological malignancy have been associated with IgA vasculitis.47–51 One study found that nearly a third of adults with IgA vasculitis purpura had an associated tumor.47 For this reason, the authors concluded that physicians should suspect an underlying malignancy in older patients (especially males of 40 years or more) with IgA vasculitis.48
Pulmonary hemorrhage is a rare complication that may prove fatal.52,53
Pathogenesis and histologic features An incomplete picture of the pathogenesis of IgA vasculitis/Henoch-Schönlein purpura has emerged. As noted above, it seems a wide variety of infective agents may trigger this disease. It is associated with IgA deposition in blood vessel walls, both in the dermis and in the renal glomerulus (mesangium). IgA1 is the major IgA subclass found in the dermal, gastrointestinal, and glomerular blood vessels.54,55 Fibrinogen and C3 are also usually present. Raised levels of serum IgA and IgE are present in some, but not all, patients.8 IgA antineutrophil cytoplasmic antibodies (ANCA) and IgA anticardiolipin antibodies have also been documented.56,57 There is evidence that patients have an increased number of IgA-type B cells.58 More recently, IgA-binding regions from streptococcal M proteins have been identified in a significant subset of skin and renal biopsies from patients with Henoch-Schönlein purpura.59
not seen in any patients in two series.35,36 Rarely, gastrointestinal involvement with minimal skin lesions is encountered.37
Renal symptoms are variable and include microscopic hematuria, acute nephritic syndrome, nephrotic syndrome, and acute or chronic renal failure. The pathological features seen on renal biopsy range from mild focal glomerulonephritis to necrotizing or proliferative glomerulonephritis.1,8 In a consecutive series of 100 pediatric patients, a single patient required transplantation.8 Patients older than 7 years are at increased risk of renal involvement.38
The finding of IgA deposition by immunofluorescence is not equivalent to a diagnosis of IgA vasculitis and must be correlated with the clinical presentation.60 A vasculitis with the presence of IgA deposition in patients lacking other typical features of Henoch-Schönlein purpura has been described in association with cancer, granulomatosis with polyangiitis, and inflammatory bowel disease.61 The observation of an association between DRB101 and DRB111 and Henoch-Schönlein purpura suggests a genetic susceptibility in some patients.62,63 Other authors have suggested that DQA1*0301 and C4 deletion may also represent risk factors for IgA nephropathy as well as Henoch-Schönlein nephritis.64 In a study from Taiwan, children with atopic dermatitis had a 1.75-fold increased risk for IgA vasculitis compared with matched controls.65
Orchitis is a recognized complication of IgA vasculitis, affecting 14% of male patients.8,25
Neurological involvement may be manifested by headaches, seizures, mental status changes, and, less frequently, ataxia and peripheral neuropathy.39–41 Neurological manifestations may be seen in up to one third of patients.41
Low serum C3, leukopenia, and thrombocytopenia are rare findings.42
IgA vasculitis in children is generally associated with a good prognosis, with less than 2% suffering long-term morbidity.43,44 However, patients do occasionally die from renal failure, gastrointestinal infarction, or respiratory involvement. In contrast to pediatric patients, adults are thought to have a worse prognosis, with remission of renal disease seen in as low as 21% of patients.45 In contrast, one study found that although older patients had more severe symptoms, including frequent renal involvement, prognosis was equally good in young and older patients.46 In another study, complete recovery was seen in 67% of adults after a median follow-up period of 36
Biopsies of cutaneous lesions show features of typical leukocytoclastic vasculitis (Fig. 16.22).
Differential diagnosis The histologic differential diagnosis includes other forms of leukocytoclastic vasculitis. Since IgA deposition can be seen in the blood vessel walls of
patients with leukocytoclastic vasculitis but without of the clinical manifestations of IgA vasculitis/Henoch-Schönlein purpura, this finding is not diagnostic in isolation.60,66 In one study, only 24% of patients with vascular IgA deposition had Henoch-Schönlein purpura.66 Other studies have demonstrated a strong correlation with vascular deposits of IgA and IgA vasculitis.5,67,68 The sensitivity and specificity has been reported to exceed 80%.5 Nevertheless, careful clinical correlation is necessary to establish the diagnosis.
723 Urticarial vasculitis
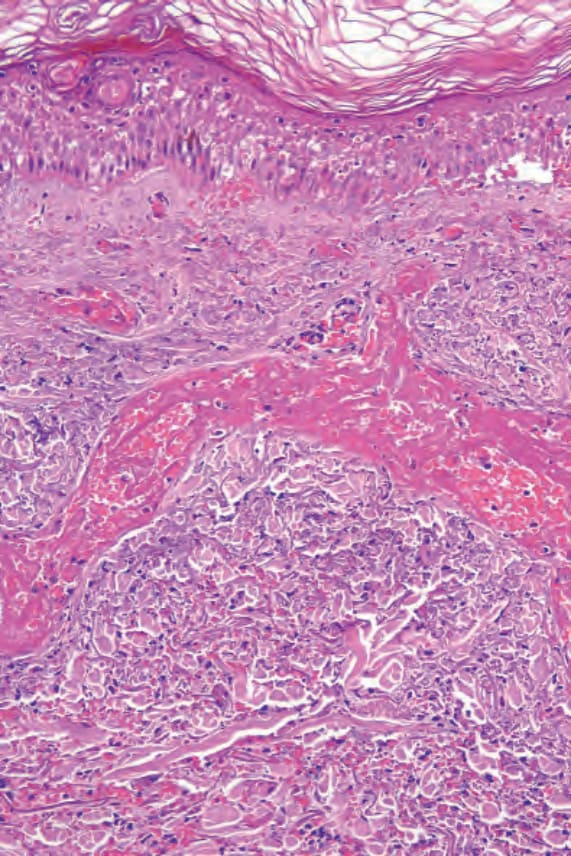
Fig. 16.19 Leukocytoclastic vasculitis: vascular thrombosis is accompanied by epidermal infarction. Note the cytoplasmic eosinophilia and loss of nuclei.
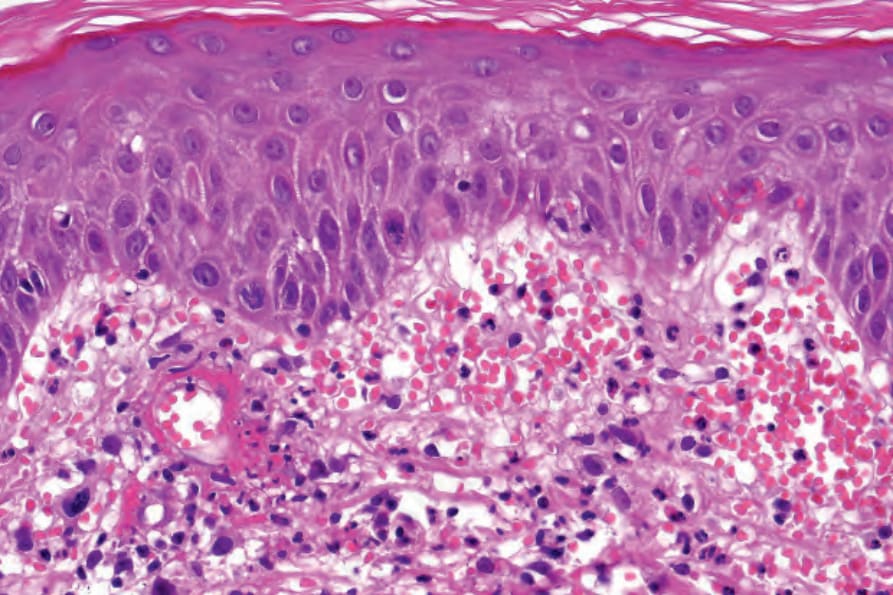
Fig. 16.20 Leukocytoclastic vasculitis: note the marked red cell extravasation.
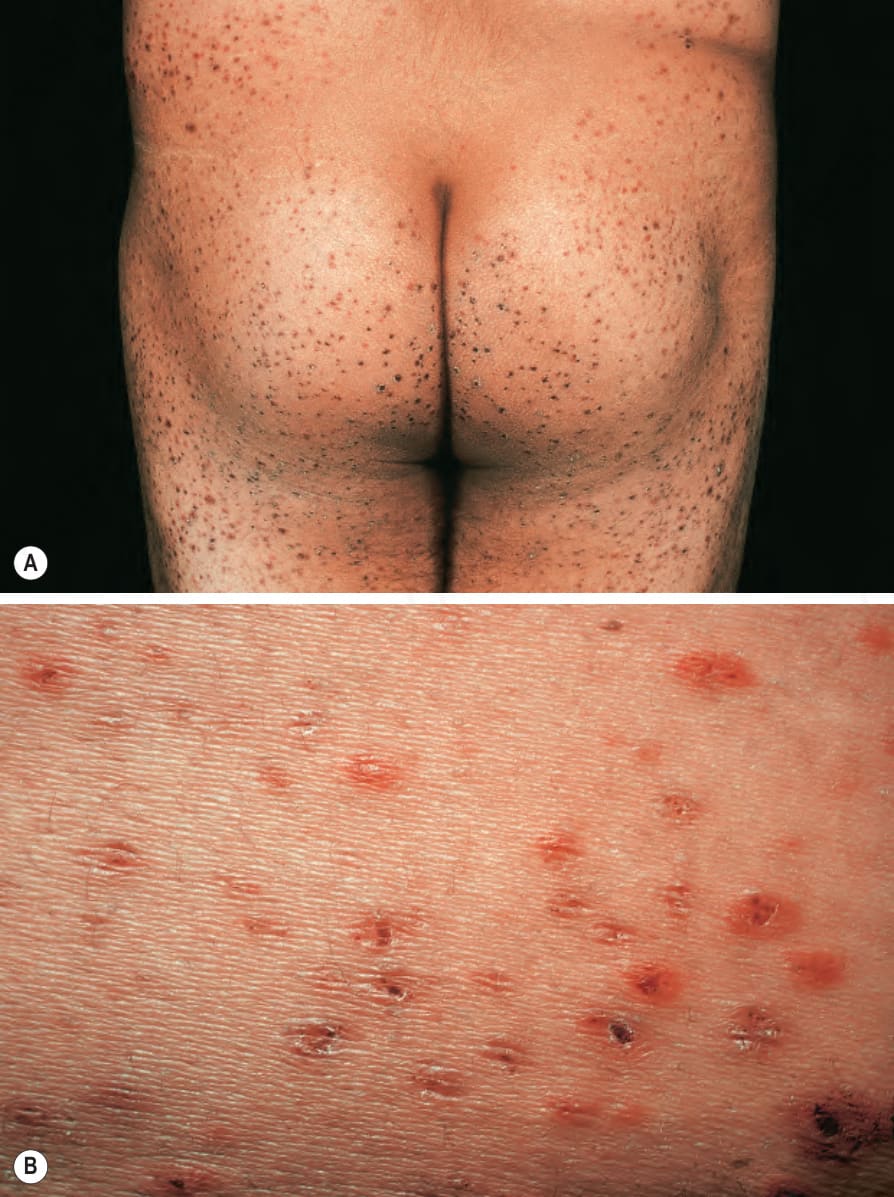
Fig. 16.21 (A, B) Henoch-Schönlein purpura: palpable purpura in the classical distribution on the buttocks and thighs. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
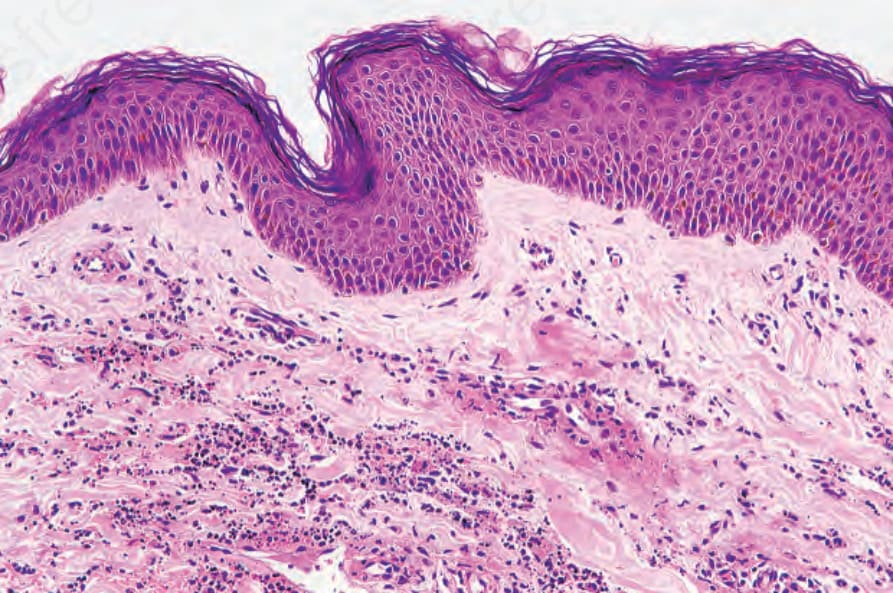
Fig. 16.22 Henoch-Schönlein purpura: this small venule shows striking fibrinoid change. There is considerable red cell extravasation.