Incontinentia pigmenti
Incontinentia pigmenti
Clinical features Incontinentia pigmenti (Bloch-Sulzberger syndrome) is a rare systemic illness with a striking female bias (in excess of 37 : 1), as affected males usually die in utero.1–3 It has an X-linked dominant mode of inheritance.4 Cutaneous manifestations usually present at birth or during the first few weeks of life.1 There may also be lesions affecting the hair, teeth, nails, eyes, skeleton, and the central nervous system in up to 80% of patients.1,4 The phenotype in females is variable, manifestations being dependent on the effects of
707 Incontinentia pigmenti
A
B
mosaicism resulting from X-chromosome lyonization.5 It has occasionally been described in identical twins.6 The disease is rarely transmitted from father to daughter.7
on the extremities (Fig. 15.58). The verrucous lesions develop at sites of previous blistering.16,17 They may resemble linear epidermal nevi.16,18 This stage (when present) appears during the second to sixth weeks of life and usually resolves completely by 6 months.5
• Stage 3, which is pathognomonic of incontinentia pigmenti, presents as bizarre reticulated pigmentation, generally between the 12th and 26th weeks after birth. It sometimes occurs de novo. The brown to slate-gray pigmentation appears as splashes, streaks, and whorls (sometimes referred to as ‘Chinese lettering’) on the torso and extremities (Fig. 15.59). The nipples are typically hyperpigmented and involvement of the groin and axillae is characteristic.5 The pigmentation appears to develop independently of the bullous lesions or verrucous plaques and follows Blaschko lines.16,17 Resolution of lesions is associated with atrophy and the pigmentation is usually imperceptible by adulthood.
• Stage 4, usually presenting in adulthood, shows only residual changes that may be visible as occasional atrophic, hypopigmented, hairless, reticulated patches and streaks best seen on the lower legs.1,5,18
Most males with incontinentia pigmenti die in utero, and only very rare patients survive. As with females, it is associated with the same phenotypic mosaicism.8 In the majority of cases of male involvement, it develops in association with Klinefelter syndrome.9–12 Half chromatid mutation, or more recently an unstable pre-mutation, has been offered as explanation for affected males with a normal karyotype.11,12 Only extraordinary cases have been reported in normal XY males, secondary to somatic mosaicism.13
Typically, the condition has four stages: 14–16
• Stage 1, comprising erythema and linear vesiculation on the trunk and extremities, is apparent at birth or during the first 2 weeks of life (Fig. 15.57).16,17 Characteristically, the face is unaffected. Patients usually show associated leukocytosis and eosinophilia. On average, the blistering stage completely resolves within 4 months.5
• Stage 2, which is uncommon and usually transitory, consists of hyperkeratotic verrucous papules and plaques most frequently found
Additional cutaneous lesions include mild nail dystrophy (40%), scalp alopecia at the vertex, and the woolly hair nevus.4,5,16 A whorled scarring alopecia following the lines of Blaschko has been documented.2,19 At this point,
708 Neutrophilic and eosinophilic dermatoses
System Abnormality
Scalp Scarring Alopecia of variable severity
Nails Occasional dystrophy
Teeth Partial/complete absence Conical (pegged)
Eyes Strabismus Blindness Cataracts Atrophy of optic nerve
Central nervous system Spastic paralysis Mental retardation Convulsions
the skin lesions cease to be problematic, and other issues such as ocular involvement are the focus of concern.20–22
Painful subungual digital verrucous nodules are occasional late features of incontinentia pigmenti.16,23–25 They are usually multiple, affect the hands more often than the feet, and, in addition to nail destruction, they can also be associated with scalloped resorption of the underlying phalanx.25 Spontaneous regression is sometimes seen. Clinically, these nodules may be mistaken for a wart, subungual fibroma, keratoacanthoma, or squamous cell carcinoma.22
Dermatoglyphic patterns presenting in patients and also in nonaffected family members have been described.26
Some patients develop episodes of late reactivation of the disease in hyperpigmented streaks, and this appears to be related to a preceding infection.27 It suggests that the mutated cells persist in the epidermis for a long period of time.
There can be widespread systemic involvement (Table 15.4). Dental abnormalities include hypodontia, delayed eruption, impaction, and crown malformations such as conical forms and accessory cusps.5,18,28–31
The most characteristic ocular changes are strabismus, microphthalmos, cataract, and optic atrophy.22,32 Retinal detachment with a fibrovascular retrolental membrane is the commonest intraocular abnormality.22,33–36
Central nervous system involvement may result in encephalopathy, seizures, mental retardation, microcephaly, cerebellar ataxia, and motor effects including spastic quadriplegia, hemiplegia, slow motor development, and psychomotor retardation.1,37–41 There is some evidence to suggest that incontinentia pigmenti can be associated with chromosomal instability and a slightly increased susceptibility to cancer; cutaneous squamous cell carcinoma has been described in a 16-year-old.42,43 A unique case associated with twenty-nail dystrophy has been reported.44
Pathogenesis and histologic features Considerable data have been published which sheds much light on the pathogenesis of this disease:
• Linkage analysis has demonstrated two incontinentia pigmenti loci that reside within the long arm of the X chromosome at Xq11 (IP1) and Xq28 (IP2).5,45,46
• Mutations in the gene for the inhibitor of kappa light polypeptide gene enhancer in B cells, kinase gamma (IKBKG) also called nuclear factor (NF)-κB gene modulator (NEMO), which plays a role in inhibiting TNF-induced apoptosis, has been shown to be responsible for development of the disease.47–50
• A mouse model has been created by disruption of NEMO, which leads to lethality in males and heterozygous females with skin and eye lesions similar to incontinentia pigmenti.51,52 Biopsy of skin lesions of diseased animals shows increased keratinocyte apoptosis, inflammation, and pigment incontinence.51
• A relationship between incontinentia pigmenti and the osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, immunodeficiency (OL-EDA-ID) syndrome has recently been suggested in a patients who
presented with the latter syndrome born to mothers with features of incontinentia pigmenti.53,54 Both diseases are associated with mutations in NEMO/IKBKG.54–56 Male patients with the disease often have less deleterious mutations and present with ectodermal dysplasia and immunodeficiency.10
• Eotaxin (an NF-kappaB-activated chemokine) is strongly expressed in the suprabasal epidermis of involved skin in patients with the disease.57 This expression is concomitant with the upper epidermal accumulation of eosinophils, suggesting a pathogenetic role for this chemokine. Histologic examination of lesions in the vesicular stage (stage 1) shows eosinophilic spongiosis (Fig. 15.60). Occasionally, aggregates of dyskeratotic cells are evident (Fig. 15.61).4 A chronic inflammatory cell infiltrate with conspicuous eosinophilia may be present within the dermis.
The verrucous lesions (stage 2) are characterized by hyperkeratosis, acanthosis, papillomatosis, and focal dyskeratosis (Fig. 15.62).24 The dyskeratotic cells are typically arranged in a whorled configuration.
Stage 3 lesions manifest marked pigmentary incontinence with numerous melanophages in the dermis associated with epidermal basal cell degeneration.
End-stage lesions display epidermal atrophy, and there may be loss of the adnexae. Melanocytes appear to be present in reduced or normal numbers
709 Hidradenitis suppurativa
A
and ultrastructurally have shown no significant lesion except for one report in which small non-dendritic forms with degenerate melanosomes were described.6,58 Some authors have recommended biopsy of these late lesions in adulthood as helpful for diagnosis.59
Molecular testing is available for confirmation of difficult cases and such techniques can also be applied to prenatal testing.3,60–62
The subungual verrucous nodules display features identical to those described in subungual keratoacanthoma with lobules of glassy cells associated with prominent hyperkeratosis, hypergranulosis, and striking dyskeratotic cells throughout the epidermis.24,25
Differential diagnosis Clinically, incontinentia pigmenti may be confused with hypomelanosis of Ito (incontinentia pigmenti achromians), and the central nervous system involvement in both diseases is similar.63D The latter condition, however, is characterized by cutaneous pigmentary changes in the absence of either vesicular or verrucous lesions.
B
Many conditions are associated with eosinophilic spongiosis, but with adequate clinical information none should pose diagnostic problems. Toxic erythema of the neonate can be distinguished histologically from incontinentia pigmenti by the absence of spongiosis in the former condition.
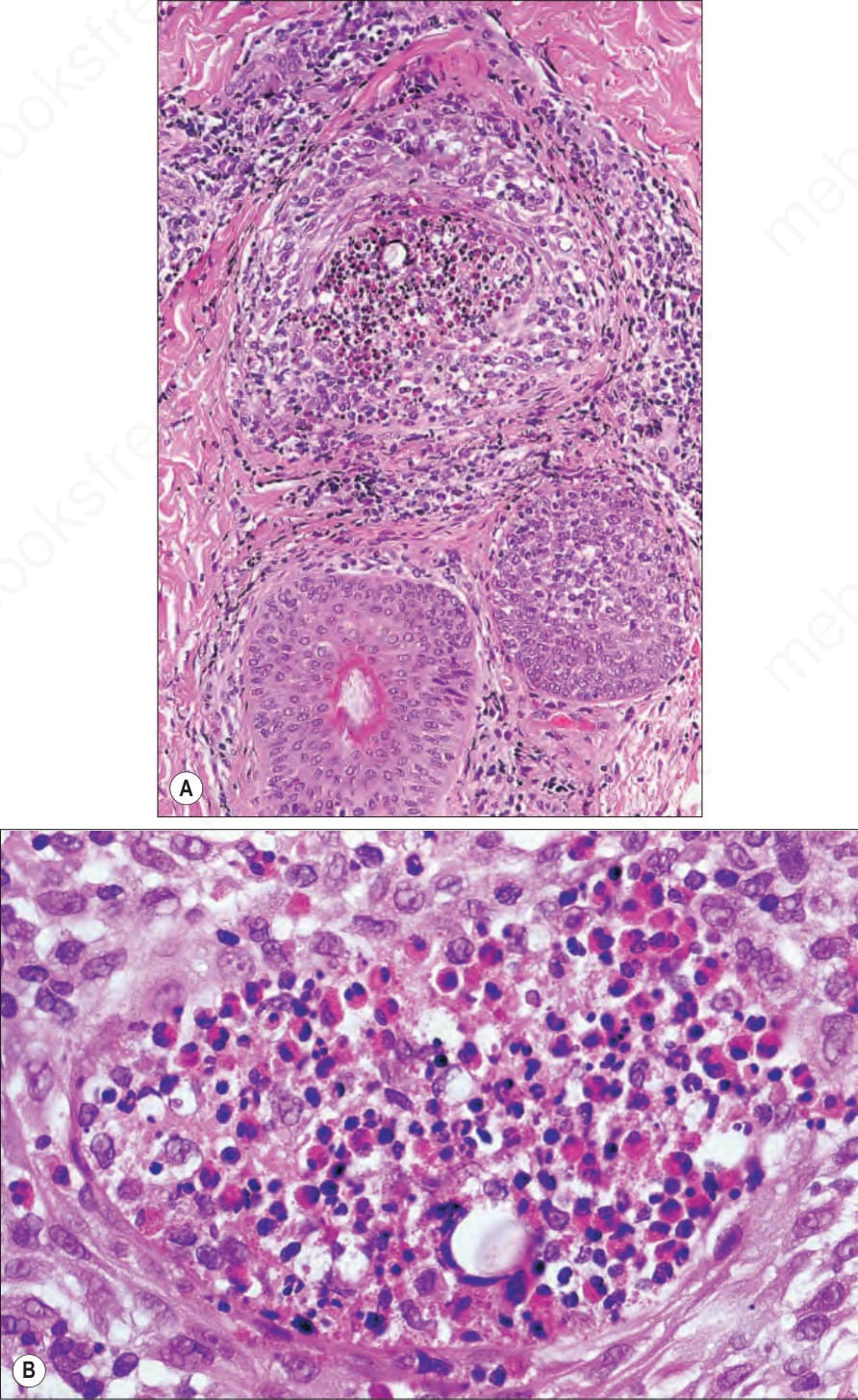
Fig. 15.57 (A, B) Eosinophilic pustular folliculitis: there is spongiosis associated with an eosinophil-rich abscess.
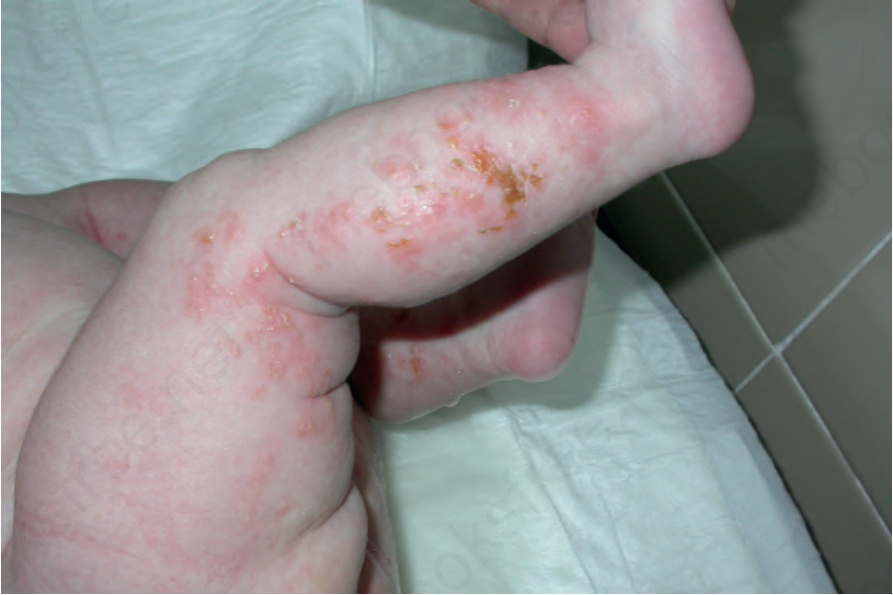
Fig. 15.58 Incontinentia pigmenti: the inflammatory stage is characterized by erythema, linear clusters of intact vesicles, crusts, and scaling. By courtesy of J.C. Pascual, MD, Alicante, Spain.
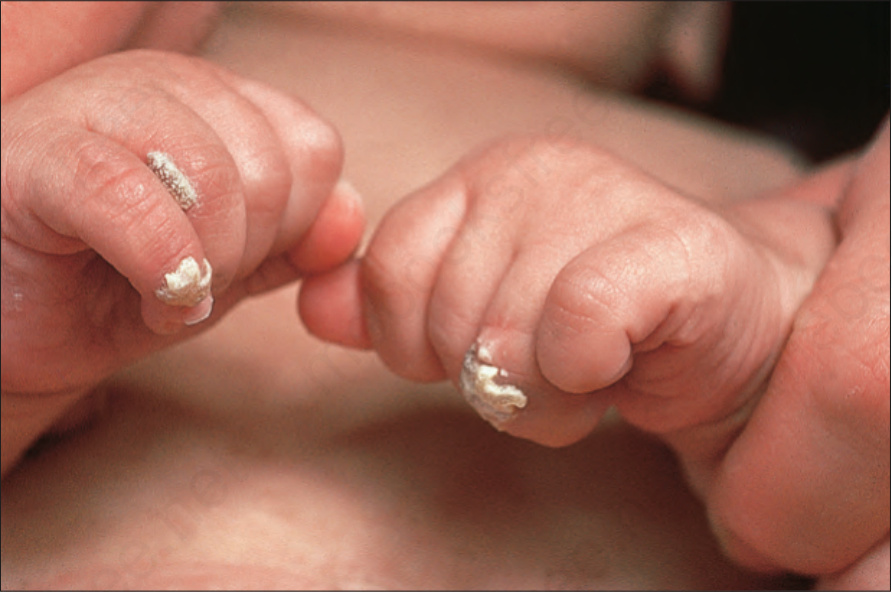
Fig. 15.59 Incontinentia pigmenti: verrucous lesions, seen in the second stage, predominantly affect the extremities. By courtesy of the Institute of Dermatology, London, UK.
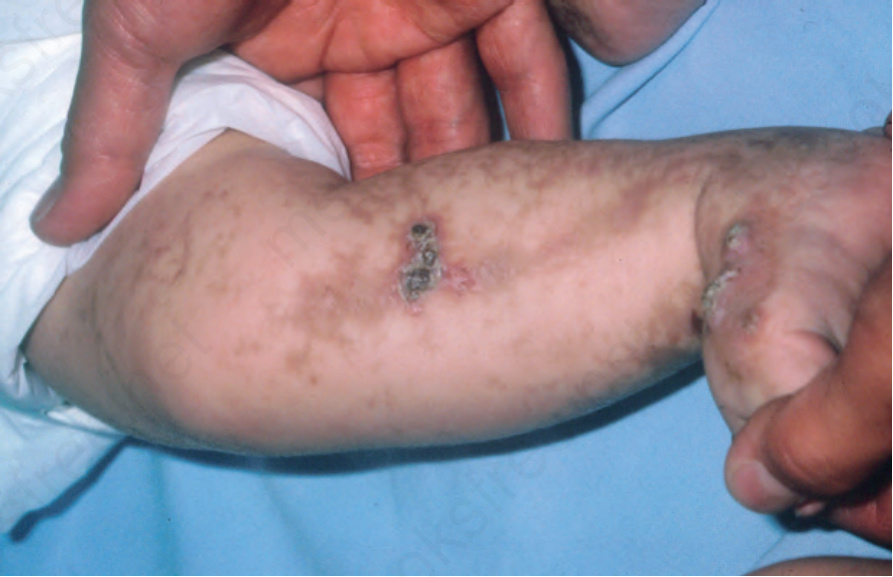
Fig. 15.60 Incontinentia pigmenti: the whorl-like distribution of the pigment is characteristic. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
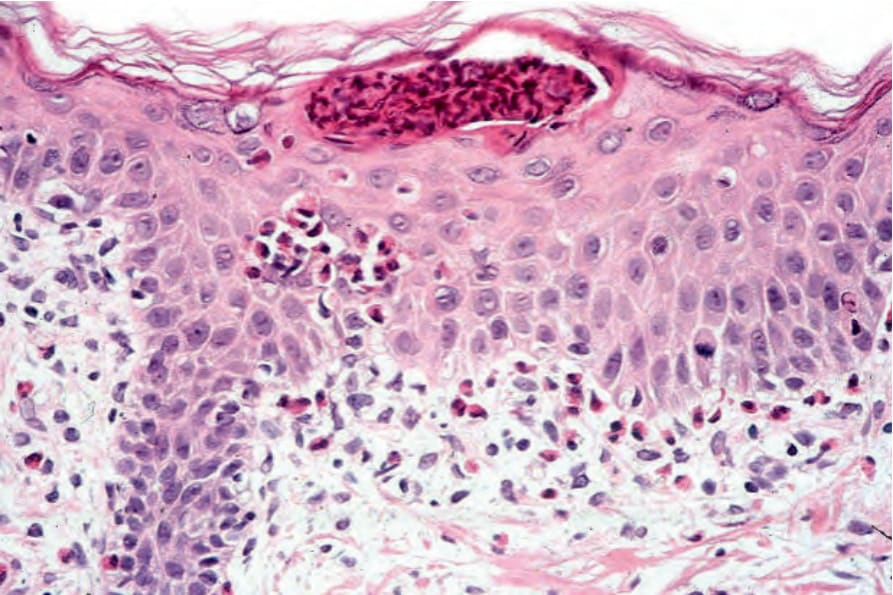
Fig. 15.61 Incontinentia pigmenti: vesicular stage showing a subcorneal eosinophil pustule and intraepidermal eosinophils.
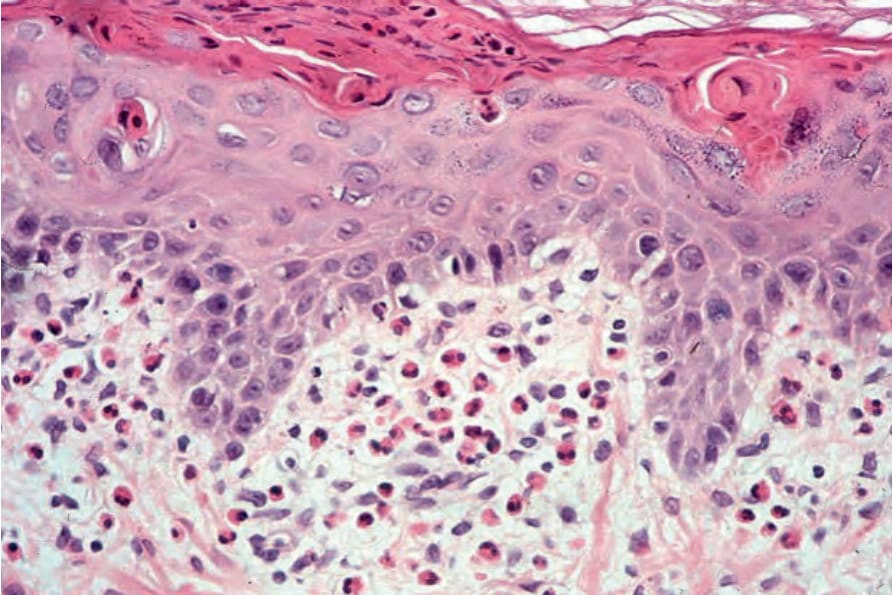
Fig. 15.62 Incontinentia pigmenti: there is dyskeratosis and an upper dermal eosinophil-rich infiltrate.
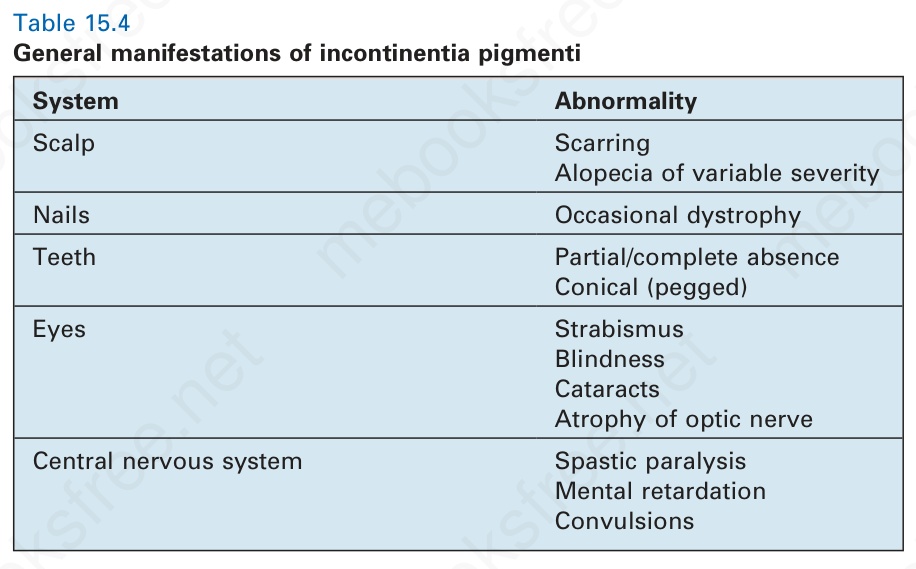
Table 15.4 General manifestations of incontinentia pigmenti