Hypereosinophilic syndrome
Hypereosinophilic syndrome
Clinical features The hypereosinophilic syndrome was previously defined as an idiopathic condition characterized by persistent eosinophilia (more than 1.5 × 109/L) for at least 6 months and with involvement of one or more organs.1–4 This definition has undergone revisions to include other diseases associated with eosinophilia such as eosinophilic pneumonia, eosinophilic gastrointestinal disorders, and eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome and discussed in the chapter on vasculitis). The definition has also been modified to include patients with peripheral eosinophils with substantial tissue eosinophilia, removing the requirement of an absolute eosinophil count > 1.5 × 109/L.5 Similarly, the time requirement of 6 months has been shortened to 1 month since some patients developed end-organ damage with less peripheral eosinophilia in a shorter period.6 The diagnosis should be made only after other causes of eosinophilia, particularly parasitic infections, have been excluded. The heart, lungs, central and peripheral nervous system, liver, and skin are commonly affected. The disease, which may sometimes prove fatal, generally presents in adults.
Cutaneous lesions are seen in over 50% of patients and usually consist of either pruritic papules and nodules or urticaria and angioedema.5,7 Rarely, skin lesions represent an initial manifestation, and in this setting, annular erythema and erythroderma have been reported.8–12 Oral and genital erosions/ulcerations are quite characteristic and can be the first manifestation of the disease.5,13,14 Other cutaneous features include livedo reticularis, cutaneous infarction, deep vein thrombosis, blisters, aquagenic pruritus, erythema gyratum repens, and Wells syndrome.5,15–21 Hypereosinophilic syndrome has been reported in association with lymphomatoid papulosis, T-cell lymphoma, myeloid neoplasms, systemic mast cell disease, SLE, and HIV infection.5,6,22–27
neutrophilic urticarial dermatosis. Although cellulitis could be histologically indistinguishable, it is clinically different.
Cutaneous atypical neutrophilic dermatosis with lipodystrophy and elevated temperature
Cutaneous atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) is a distinctive, rare, autoinflammatory disease previously described as Nakajo-Nishimura disease, joint contractures, muscle atrophy, microcytic anemia and panniculitis induced lipodystrophy (JMP), or Japanese autoinflammatory syndrome with lipodystrophy.1–3
The condition presents early in life with recurrent, almost daily episodes of fever poorly responsive to non-steroidal anti-inflammatory drugs, purpuric, annular, violaceous skin plaques, low height and weight, eyelid swelling, lipodystrophy, increased ESR and CRP, hypochromic anemia, and elevated liver enzymes. Other less common manifestations include perioral swelling,
Pathogenesis and histologic features The etiology is unknown. It has sometimes been categorized into:
• idiopathic,
• clonal (primary clonal expansion of eosinophils),
• familial (autosomal dominant),
• secondary types.5,6,28
704 Neutrophilic and eosinophilic dermatoses
Some forms of clonal eosinophilia best regarded as variants of eosinophilic leukemia, (malignancy-associated) harbor a distinctive FIP1L1-PDGFRA fusion (FIP1L1: Fip1-like; PDGFRA: platelet-derived growth factor receptor A) and are amenable to targeted therapies.29–31 Secondary cases result from other causes such as an underlying malignancy particularly lymphoid neoplasms. The lymphoid or lymphocytic variant of hypereosinophilic syndrome results from circulating T lymphocytes that are usually CD3-CD4+, are frequently clonal and drive a polyclonal proliferation of eosinophils.32 The latter is the result of production of IL-5 by the clonal T cells. This variant of the disease may be very rarely complicated by the development of T-cell lymphoma, cutaneous or systemic. Idiopathic is what remains when these first two categories are excluded. Besides elevated serum levels of IL-5, elevated serum levels of IL-10 and soluble IL-2 receptor have also been documented.33 IL-2 stimulates release of eosinophilic cationic protein from eosinophils which contain IL-2 receptor (CD25), and this results in tissue damage.
The histologic findings vary according to the type of lesion biopsied. Urticarial and papular lesions show a superficial and deep perivascular and interstitial mixed inflammatory cell infiltrate with variable numbers of eosinophils and scattered lymphocytes, histiocytes, and occasional plasma cells. Rare flame figures may be present and may occasionally be prominent. Dermal edema is seen particularly in urticarial lesions. Eosinophils are not always prominent, and the findings can be entirely non-specific. Microthrombi are present in some cases and sometimes correlate with the severity of the disease.15,34 In the lymphocytic variant of the disease an exceptional case of cutaneous lesions with a CD30 positive clonal T-cell lymphoid proliferation has been described.35
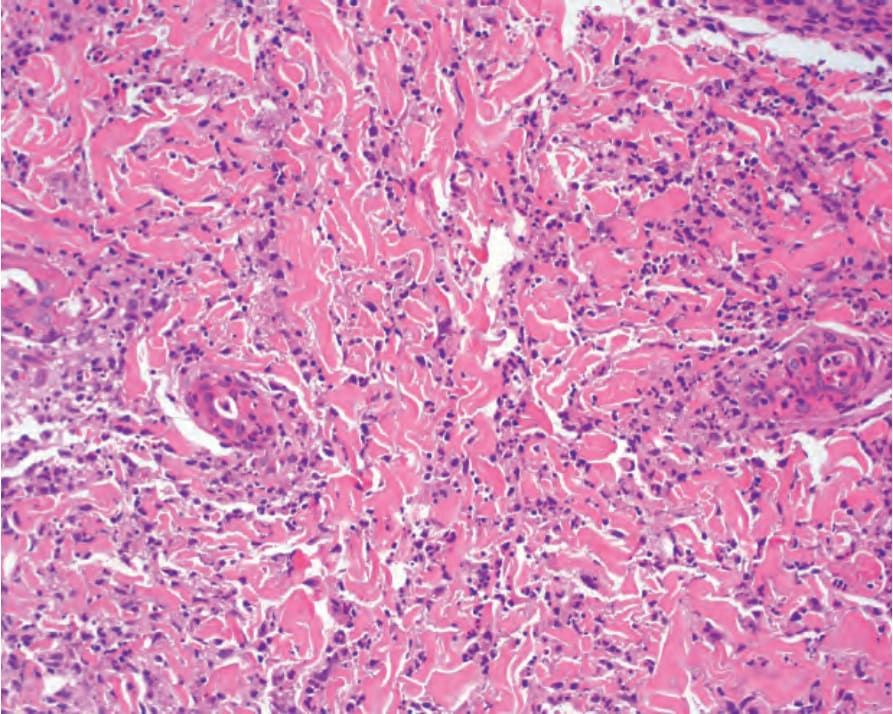
Fig. 15.48 Biopsy from patient with neutrophilic urticarial dermatitis with interstitial infiltrate with cordlike pattern intercalating between collagen bundles.
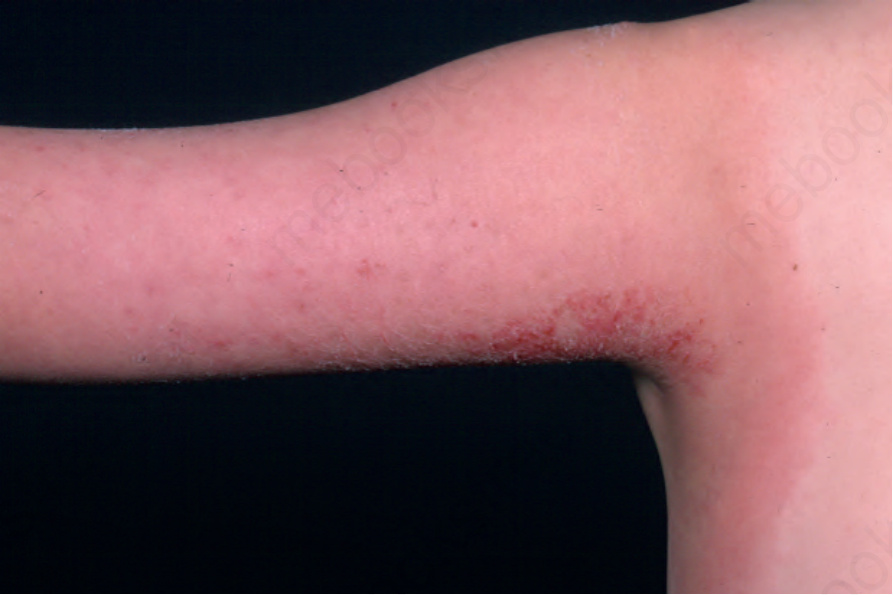
Fig. 15.49 Eosinophilic cellulitis: there is a large erythematous swollen plaque. The limbs are commonly affected. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.