Erythema marginatum rheumaticum
Erythema marginatum rheumaticum
Clinical features Once a common disease, it was thought that, with the effective antibiotic treatment of the causative infection, rheumatic fever would become of historical interest only. However, there has been a resurgence of the condition over the past few decades, particularly in developing countries.1–5
Rheumatic fever is an immunologically mediated disease that follows an infection with Lancefield group A beta-hemolytic streptococcus. The infection causes pharyngitis and carditis. Additional features include polyarthritis, a neurological movement disorder known as Sydenham chorea, and subcutaneous nodules.6 Carditis, characterized by a valvular disease, is a major cause of morbidity and mortality.
Juvenile rheumatoid arthritis is classified into three variants: pauciarticular, polyarticular, and systemic onset.
• The pauciarticular (oligoarticular) form is characterized by arthritis involving up to a maximum of four joints. Systemic manifestations are uncommon. Uveitis, however, is frequently present.
• The polyarticular form is manifest by symmetrical arthritis typically involving the knees, wrists, and ankles. Fever and hepatosplenomegaly are sometimes present.
• The systemic-onset form is a severe variant in which lymphadenopathy, fever, and rash precede development of polyarteritis, which most often affects the knees, ankles, and wrists.9 Additional features may include hepatosplenomegaly and effusions. In general, the term Still disease is restricted to this form of juvenile rheumatoid arthritis; however, some authors use it for any of the variants. The majority of patients with the systemic form have the characteristic rash in contrast to the pauci- and polyarticular variants in which only 20–40% are affected.9
Erythema marginatum rheumaticum is the designation given to the distinctive annular or polycyclic eruption of rheumatic fever. The lesions are nonpruritic, multiple, flat, erythematous maculopapules which change and spread over hours, and are often recurrent. The trunk and proximal extremities are most frequently affected.2,7 The hands and face may also be involved.4 By definition, erythema marginatum rheumaticum is associated with rheumatic fever, but occurs in only 1–18% of patients.1 Some studies have failed to identify significant HLA associations.3,8 However, there are conflicting reports of certain HLA subtypes and the disease.9
Pathogenesis and histologic features The pathogenesis of rheumatic fever is incompletely understood. It appears likely that it results from a hypersensitivity reaction triggered by streptococcal infection. Specifically, patients develop autoantibodies that cross-react
The rash of Still disease is typically evanescent, and is characterized by a faint erythematous (salmon-colored), sometimes pruritic, macular eruption involving the trunk, extremities, head, and neck.9–12 Often, there is an association between onset of rash and febrile episodes, particularly in the late afternoon or evening.9,11 The rash is typically present for only a short period, usually a matter of a few hours; however, some lesions persist for more than 24 hours.9,11,12 It characteristically reappears without regard for its former distribution.11 Macules are often only a few millimeters in size but frequently become confluent to form larger lesions. Central pallor is sometimes a feature of the latter.10 The eruption – which may persist for weeks to years – tends to localize to areas of mild trauma and pressure.9,11
Laboratory abnormalities include elevated ESR and C-reactive protein. Serum immunoglobulins may also be raised. Patients often have leukocytosis
697 Urticaria
and increased ferritin levels and may have anemia, and thrombocytosis.12 Occasional patients have rheumatoid factor, and some authors consider this to represent bona fide juvenile rheumatoid arthritis. Antinuclear antibodies are commonly found in patients with pauci- and polyarticular variants of the disease.8 In contrast, antinuclear antibody is usually not present in the systemic-onset form.
It is very difficult to predict the outcome of this disease in the individual patient. Approximately 50% of patients experience symptoms into adulthood. Progression of juvenile rheumatoid arthritis to SLE has been documented.13 Serious complications including uveitis, cardiac tamponade, portal vein thrombosis, liver failure, disseminated intravascular coagulation, parenchymal lung disease, and hemophagocytic syndrome have been documented.14–22
Despite the name juvenile rheumatoid arthritis, Still disease is not limited to the pediatric population. Adult onset is well described in the literature (Fig. 15.36).12,23–29 Patients with the adult form of the disease may develop persistent papules and plaques and hyperpigmentation.30 Lesions are mainly seen on the face, neck, trunk, and extensor surfaces of the extremities.
The influence of various HLA alleles and their association with juvenile rheumatoid arthritis has been an area of considerable interest and has yielded a complex picture of the relationship between certain HLA alleles and risk of disease.37 HLA-A2, DR8, DR5, and DPB1*0201 are associated with increased risk of pauciarticular disease early in life.37 While B27 and DR4 may be protective in the early years, these alleles seem to confer increased risk of disease later in life.38
CD4-reactive T lymphocytes are the predominant cell type in the inflamed synovium.39 As with other autoimmune disorders, production of predominantly Th1 cytokines (IFN-γ and IFN-β) has been observed in the synovium of juvenile rheumatoid arthritis patients.39,40
Pathogenesis and histologic features The pathogenesis of juvenile rheumatoid arthritis is poorly understood. It would seem likely, however, that the various subtypes have different etiologies. Thus:
• IL-2 mRNA is detected more often in pauciarticular juvenile rheumatoid arthritis than in the polyarticular form.31
• A shift toward a Th1 type cytokine profile is seen with increased serum levels of IFN-γ, IL-6, IL-18, and TNF-α.28,31,32
• IL-18 may play a particularly crucial role in activating macrophages and inducing the TH1 cytokine profile in Still disease.28
The biopsy findings are non-specific and variable. There is often a perivascular neutrophilic infiltrate.10,12 In some cases, mononuclear cells are the predominant cell type, and in others there is a mixed infiltrate of neutrophils and mononuclear cells.9,11,12 A neutrophilic panniculitis may be associated with the disease.41 In adult-onset Still disease, distinctive histologic features have been described particularly in the persistent papules and plaques sometimes seen in the disease. The epidermis displays dyskeratotic keratinocytes in single units or aggregates (Fig. 15.37).12,30 They tend to be present mainly in the upper layers of the epidermis and even in the stratum corneum.12,30,42 Other changes include subcorneal pustules, upper dermal lymphocytes and neutrophils, interface change, neutrophilic eccrine hidradenitis, and increased dermal mucin.12,30,42
Differential diagnosis The histologic findings, as indicated above, are variable and non-specific. Clinical correlation is necessary to establish the diagnosis. The differential diagnosis includes infection, and culture and stains for microorganisms should be performed when necessary. The absence of fibrinoid change and necrosis of blood vessel walls distinguishes the lesions from leukocytoclastic vasculitis. The presence of dyskeratotic cells could suggest an irritant contact dermatitis, but that entity has less inflammation and a different clinical presentation.
It has been suggested that nucleotide-binding and oligomerization domain (NOD)-like receptors (NLR) that detect microbes and help constitute the inflammasome (a complex of proteins which initiates an inflammatory reaction) may be involved in autoimmune disease generally and in Still disease in particular.33,34
Data suggesting that the incidence of the disease is decreasing with cyclical peaks raise the possibility that environmental factors could play a role in the pathogenesis.4 Demonstration of T-cell oligoclonal expansions within synovial tissue suggests that an antigen or group of antigens may trigger the disease.35 The nature of such triggering factors, however, has not yet been identified.
The prevalence of autoimmune disease is increased in relatives of patients with juvenile rheumatoid arthritis compared with control subjects, suggesting that shared susceptibility genes may be of importance in the pathogenesis of juvenile rheumatoid arthritis and other autoimmune diseases.36
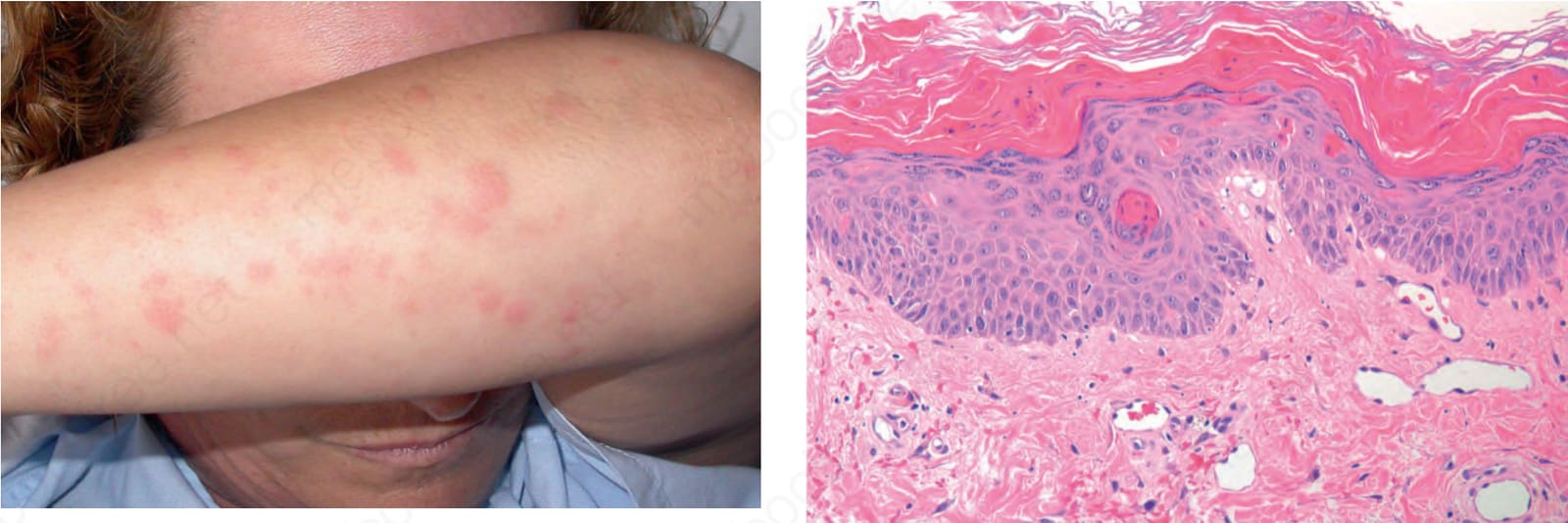
Fig. 15.36 Still disease in an adult: there are multiple erythematous macules. By courtesy of J.C. Pascual, MD, Alicante, Spain.
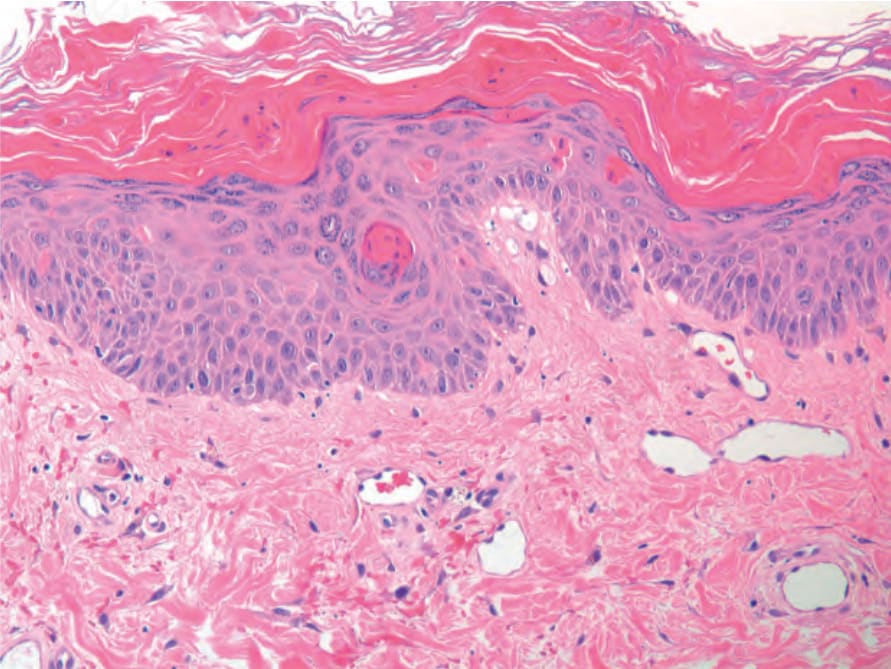
Fig. 15-37 (caption embedded in image / 圖說烘焙於圖內)