Variegate porphyria
Variegate porphyria
Clinical features This familial type of porphyria manifests the cutaneous features of porphyria cutanea tarda and the acute abdominal and neurological attacks of acute intermittent porphyria, both of which usually become apparent in the second or third decade (Figs 13.106–13.109).1,2 It is particularly common in South Africa where it can be traced to the descendants of a single Dutch family.2,3 It is an autosomal dominantly inherited condition and more severely affected homozygotes have been recognized (Figs 13.110 and 13.111). Variegate porphyria is associated with diminished activity of protoporphyrinogen oxidase, the penultimate enzyme in the heme biosynthetic pathway.4 Several different mutations have been demonstrated in the protoporphyrin oxidase gene on chromosome 1q22-23.5–7 The genotype is not a significant determinant of the clinical manifestations.8–10 Usually, there is approximately a 50% reduction in the activity of the enzyme.10
some of the mutations that have been described.2 This form of porphyria is also heterogeneous and different mutations in the UROD gene may occur.3–7 The activity of uroporphyrinogen decarboxylase is much lower than in porphyria cutanea tarda. As a consequence, disease manifestations are typically severe. Mild variants have been reported in association with certain genetic
Acute attacks may be precipitated by a wide range of drugs that induce hepatic microsomal activity, including barbiturates, alcohol, oral contraceptives, pregnancy, anticonvulsants, and sulfonamides.11–14 Acute variegate porphyria has also presented during an episode of viral hepatitis.11 The cutaneous manifestations are sometimes mild or absent during the acute attack and the condition may therefore be misdiagnosed as acute intermittent porphyria.2
Histologic features of the porphyrias Direct immunofluorescence reveals immunoglobulin (particularly IgG and to a lesser extent IgM), fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis (Fig. 13.112). Although
596 Degenerative and metabolic diseases
this is particularly evident in erythropoietic protoporphyria, it is also a feature of the other ‘cutaneous’ variants.1–3 Immunoreactants are also frequently present at the dermal–epidermal junction and have been identified within the basement membrane region of eccrine sweat glands and ducts.1–5 This finding is believed to be due to the non-specific binding of serum components rather than an immunologically mediated reaction.4 In addition, both type IV collagen and laminin are present in increased amounts, thereby contributing to the vessel wall thickening.6 Cytoid bodies are also commonly evident.4 Direct immunofluorescence studies have demonstrated granular and homogenous deposition of the membrane attack complex C5b-9 in vessel walls of the superficial and mid dermis in patients with porphyria cutanea tarda. It is proposed that UV light activated uroporphyrins
597 Porphyria
A
in turn activate complement, possibly playing a pathological role.7 Indirect immunofluorescence is invariably negative for basement membrane zone autoantibodies.
The histologic changes for all types of porphyria are very similar. The characteristic feature is the presence of a PAS-positive, diastase-resistant, hyaline material around the blood vessels of affected skin (Figs 13.113 and 13.114). In mild disease the deposits are delicate and are usually limited to the papillary dermal blood vessels, but in more severe cases the deposits are widespread, occur more deeply in the dermis, and give the vessel walls a characteristic lamellated appearance. These appearances are particularly conspicuous in erythropoietic protoporphyria.1,8 Alcian blue-positive mucin is sometimes evident around the blood vessels and to a lesser extent at the dermal–epidermal junction in both porphyria cutanea tarda and erythropoietic protoporphyria.1 Lipid droplets are sometimes also demonstrable. A false-positive Congo red stain for amyloid may be evident in the lower dermis.1
B
Electron microscopic observations include considerable basement membrane reduplication around the dermal vasculature and to a lesser extent at the dermal–epidermal junction (Fig. 13.115).1,8 This is consistent with the effects of repetitive endothelial cell injury and regeneration with subsequent new basement membrane formation. In addition, finely fibrillar material is typically present both around the vessels and at the epidermal basement membrane region. Irregular electron-dense amorphous deposits may also be evident.1
There may be subepidermal blisters, characteristically associated with slight mononuclear inflammatory cell infiltration (Figs 13.116 and 13.117). Neutrophil polymorphs showing leukocytoclasis have been described in acute lesions of erythropoietic protoporphyria and red cell extravasation is sometimes evident.9 Festooning of the dermal papillae is often, though not invariably, present. The plane of cleavage appears to be variable.3,10 Some blisters arise beneath the lamina densa in the superficial dermis similar to epidermolysis bullosa acquisita. In others, they develop within the reduplicated basement membrane constituents. Most often, however, as shown by antigen mapping experiments, blistering commences in the lamina lucida.10,11 Type IV collagen and laminin are therefore usually present along the floor of the blister while bullous pemphigoid antigen is evident in the roof. Linear segmented structures composed of type IV collagen and laminin have been identified in the roof of blisters from patients with porphyria cutanea tarda.3,12 These so-called caterpillar bodies may also be seen in specimens from patients with erythropoietic protoporphyria and drug-induced pseudoporphyria (Fig. 13.117b).13 They are PAS positive and appear as globules arranged in a linear fashion in the epidermis overlying the subepidermal blisters. Ultrastructural studies suggest that these bodies represent a combination of degenerating keratinocytes, colloid bodies, and basement
membrane fragments formed by repeated blistering and re-epithelialization.14 Caterpillar bodies are present in up to 43% of porphyria cutanea tarda specimens.15 ‘Caterpillar body-like clusters’ have also been identified in patients with porphyria cutanea tarda, erythropoietic protoporphyria, bullous pemphigoid, and junctional and dystrophic epidermolysis bullosa. These clusters were histologically identical to classical caterpillar bodies, but did not stain for type IV collagen or PAS stains.15
Rarely, a lichenoid tissue reaction has been documented in porphyria cutanea tarda.16
The histologic features of the blisters seen in variegate porphyria are identical to those described for porphyria cutanea tarda.17
A secondary sclerodermatous change is frequently present in more chronic lesions, characterized by thickened collagen bundles and reduced numbers of cutaneous adnexae (Fig. 13.118).4,8,18 Diastase-resistant, PAS-positive material may be identified throughout the involved dermis.1 This is particularly marked in porphyria cutanea tarda. Its distinction from the dermal changes of scleroderma may be very difficult, but it has been said that the texture of the collagen bundles is somewhat looser in porphyria.1 The clinical distribution of sclerodermoid changes in porphyrias in sun-exposed skin aids in the distinction.19 Basement membrane thickening due to diastase-resistant, PAS-positive material is usually present, particularly in porphyria cutanea tarda.1,8 Solar elastosis is often evident in the latter condition, but this is probably largely a consequence of the age of the patient and is unlikely to be a fundamental process (Fig. 13.119). It is not a feature of erythropoietic protoporphyria.1,8
598 Degenerative and metabolic diseases
A
In the alopecia associated with porphyria cutanea tarda, the initial changes are those of swelling and homogenization of the perifollicular connective tissue sheath.20 Later, the features of sclerodermatous transformation of the reticular dermis supervene. Centrofacial papular lymphangiectasis is characterized by the presence of dilated lymphatics in the superficial dermis.21
The hepatic changes of porphyria cutanea tarda are variable and include needle-shaped uroporphyrin crystals, hepatitis, liver cell degeneration and regeneration, fatty change, hemosiderosis, and scarring, sometimes amounting to cirrhosis (Fig. 13.120).20,22,23 There is an increased risk of hepatocellular carcinoma.23–27 Hepatic changes of erythropoietic protoporphyria include birefringent, dark brown, protoporphyrin crystal deposition in the
B
599 Pseudoporphyria
hepatocytes and Kupffer cells, hepatocyte necrosis, portal and periportal fibrosis, cholestasis and, less commonly, cirrhosis (Fig. 13.121).28
Differential diagnosis The major differential diagnosis histologically is between porphyria, pseudoporphyria, epidermolysis bullosa acquisita and congenita, and bullous amyloidosis. All produce cell-poor or cell-free subepidermal blisters. Their distinction is readily made in the majority of cases with clinical information, immunofluorescence studies, and Congo red staining.
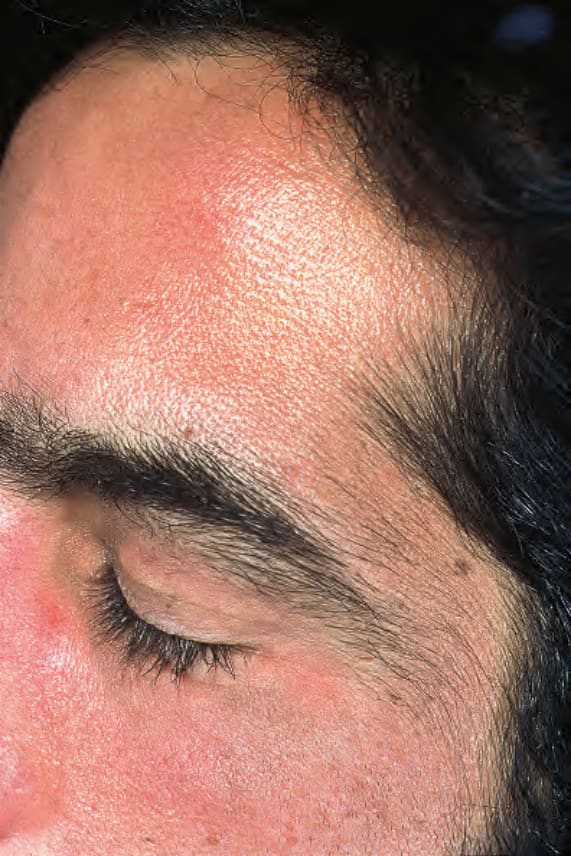
Fig. 13.105 Porphyria cutanea tarda: hypertrichosis as seen in this patient is a very typical feature. By courtesy of the Institute of Dermatology, London, UK.
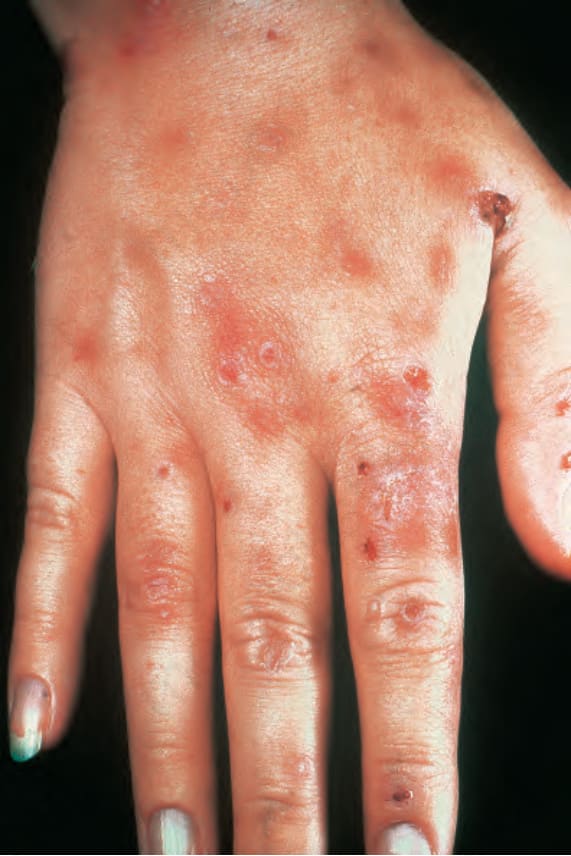
Fig. 13.106 Variegate porphyria: numerous ruptured vesicles are present on the back of the hand and fingers. By courtesy of G. Murphy, MD, Beaumont Hospital, Dublin, Eire.
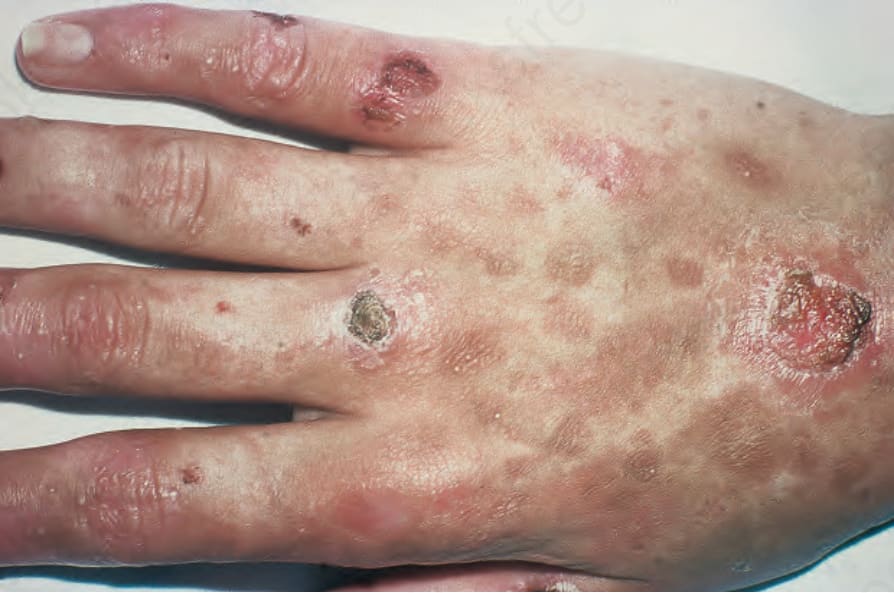
Fig. 13.107 Variegate porphyria: there are ruptured blisters with scarring and milia. By courtesy of the Institute of Dermatology, London, UK.
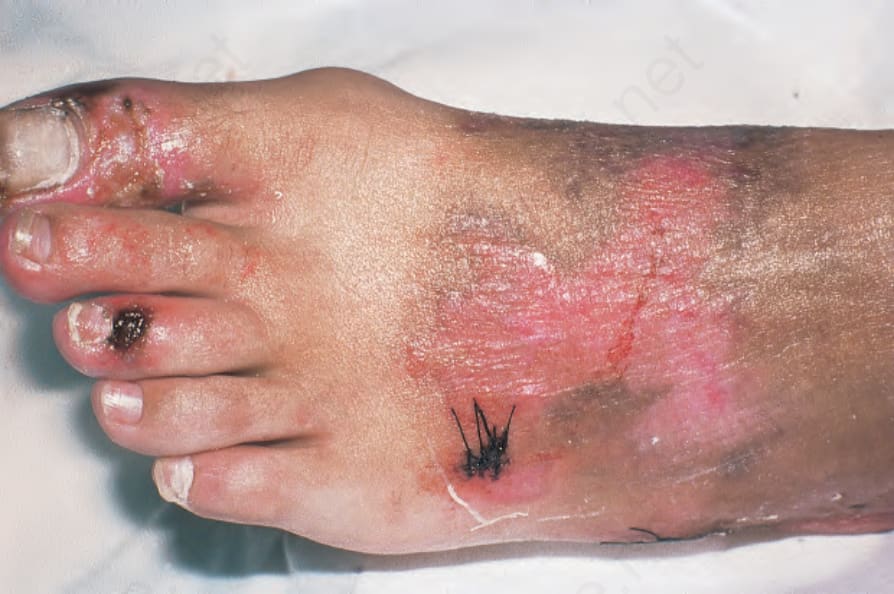
Fig. 13.108 Variegate porphyria: note the blistering over the toes and dorsum of the foot. By courtesy of the Institute of Dermatology, London, UK.
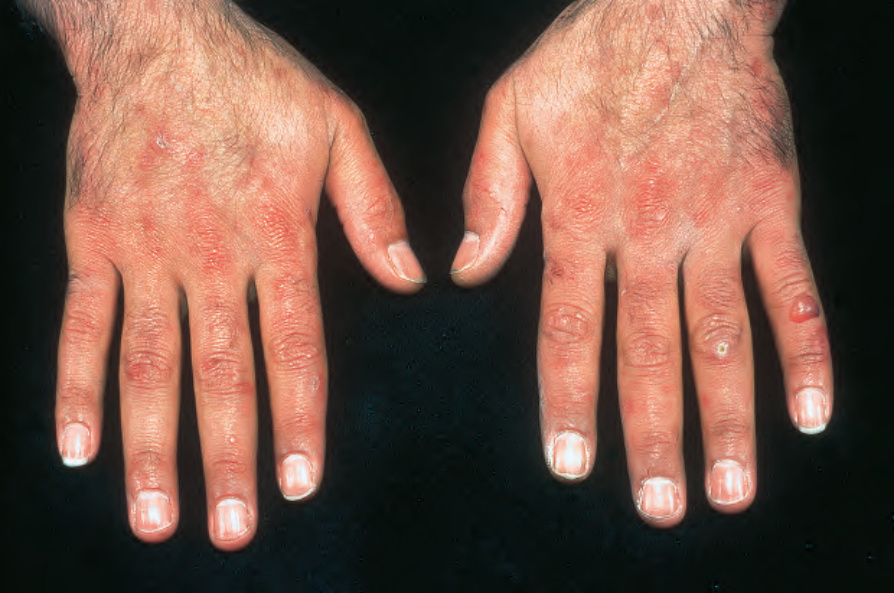
Fig. 13.109 Variegate porphyria: an intact blister is present on the left little finger. Elsewhere, there is marked scarring and milia are present. By courtesy of the Institute of Dermatology, London, UK.
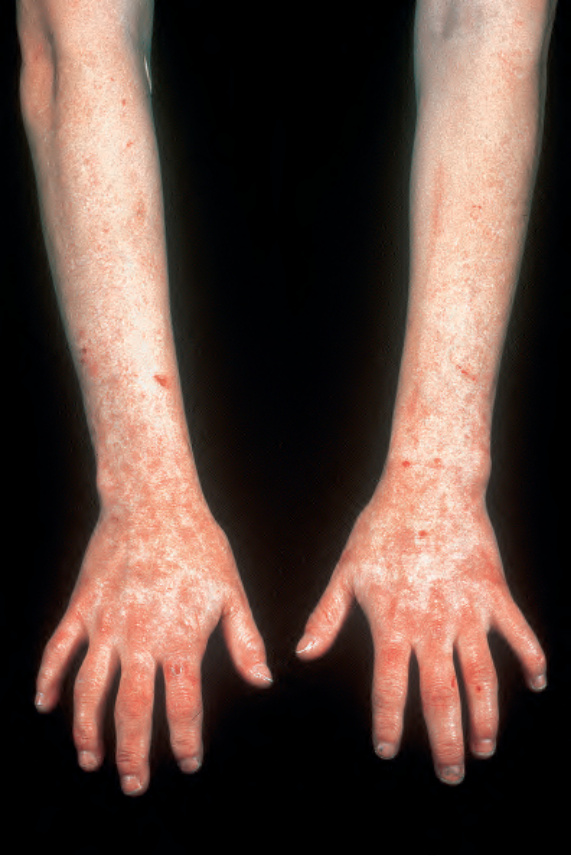
Fig. 13.110 Homozygous variegate porphyria: there is marked scarring of the dorsal surface of the forearms, hands, and fingers. By courtesy of the Institute of Dermatology, London, UK.
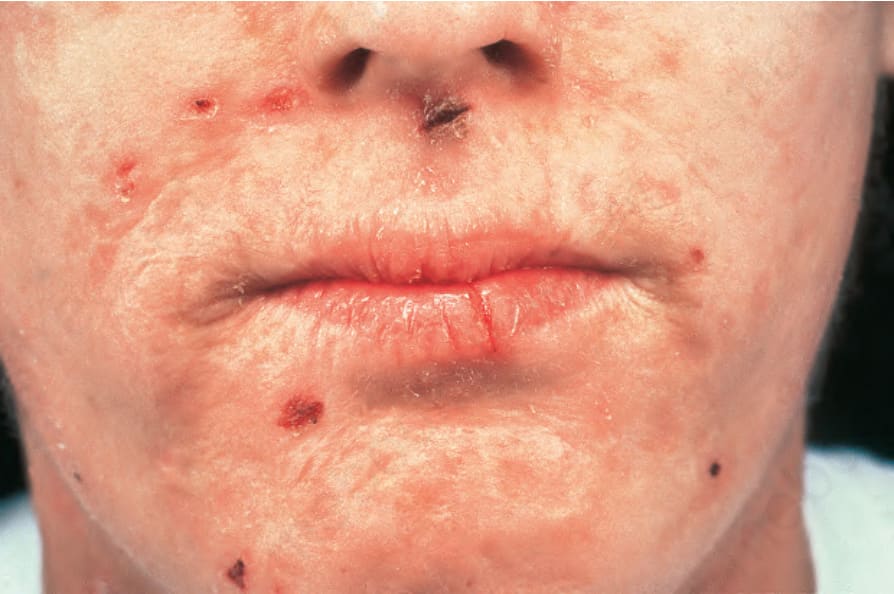
Fig. 13.111 Homozygous variegate porphyria: note the perioral erosions and scarring. By courtesy of the Institute of Dermatology, London, UK.
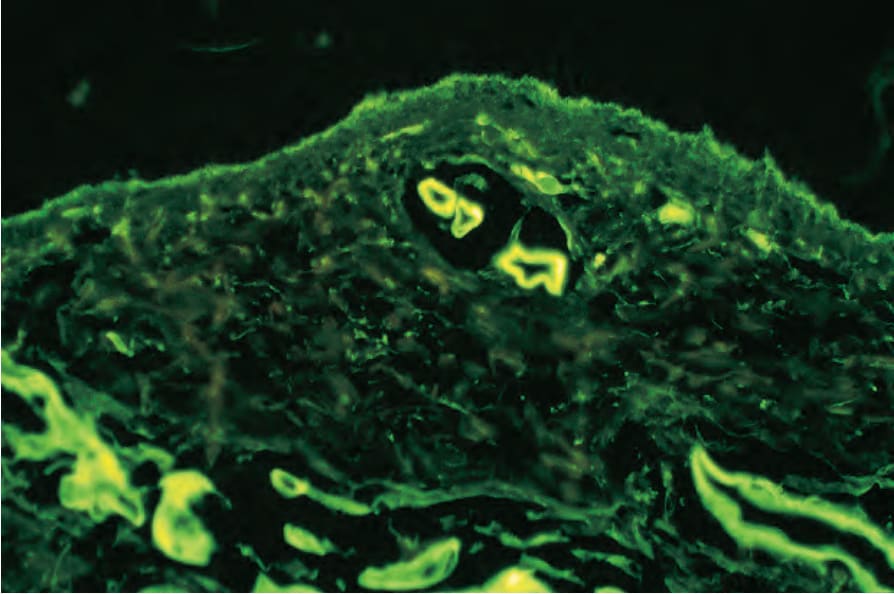
Fig. 13.112 Porphyria cutanea tarda: the superficial blood vessels show striking IgG circumferential deposition.
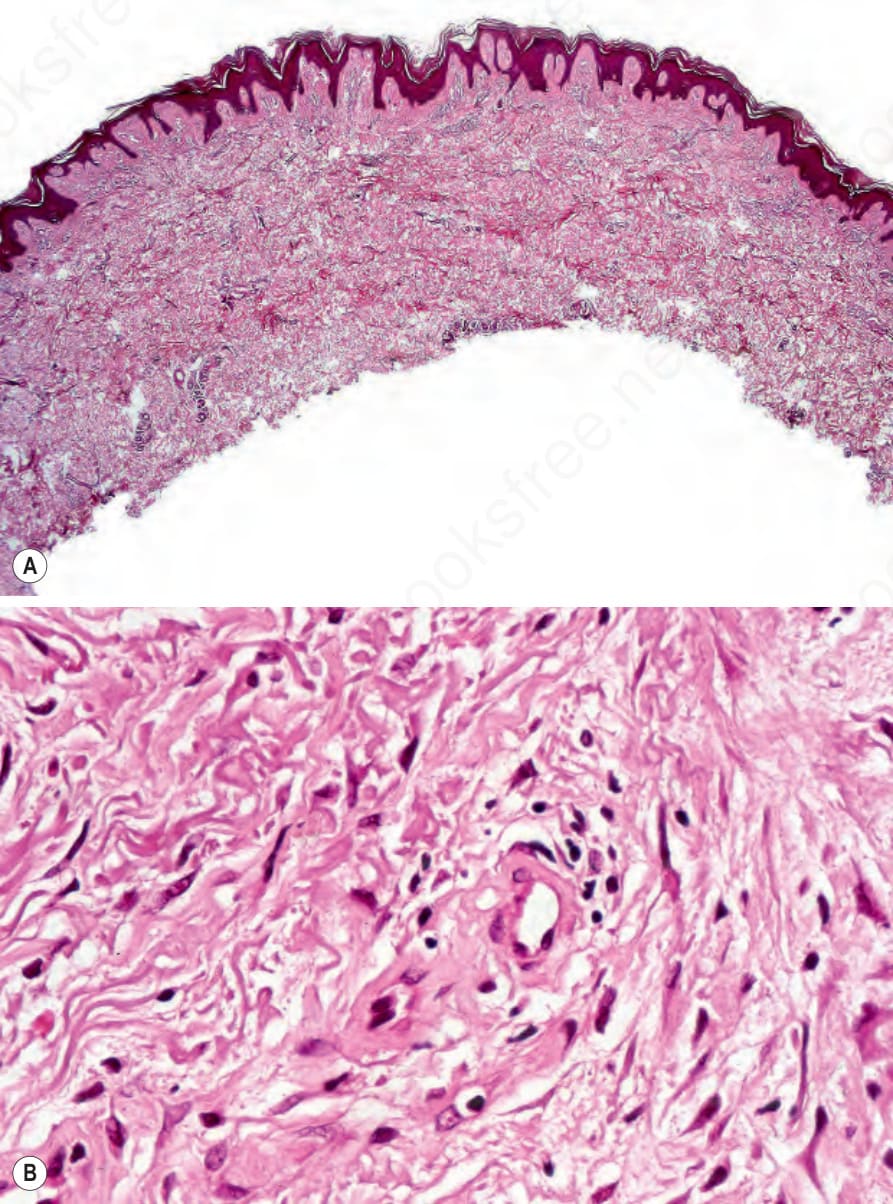
Fig. 13.113 (A, B) Porphyria cutanea tarda: the superficial vessels are thickened and appear hyalinized.
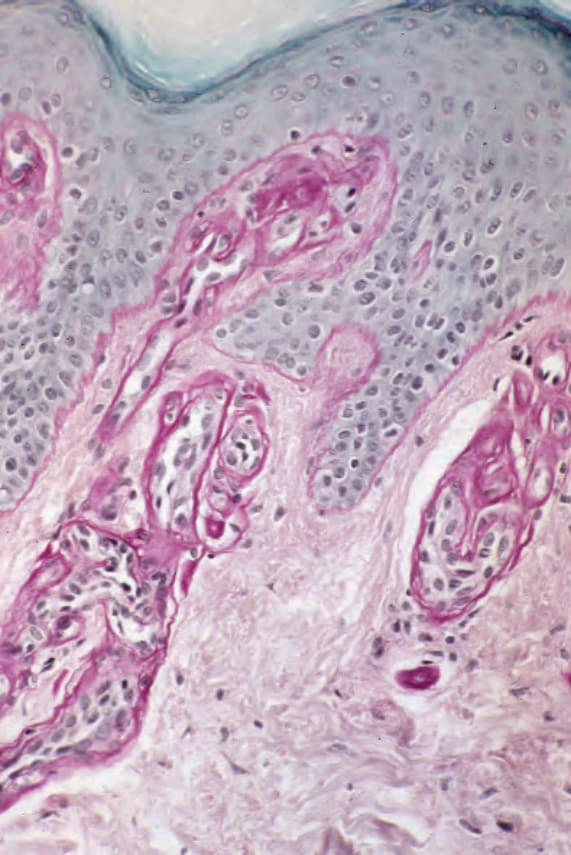
Fig. 13.114 Erythropoietic protoporphyria: the appearances are much more dramatic in this periodic acid–Schiff-stained section.
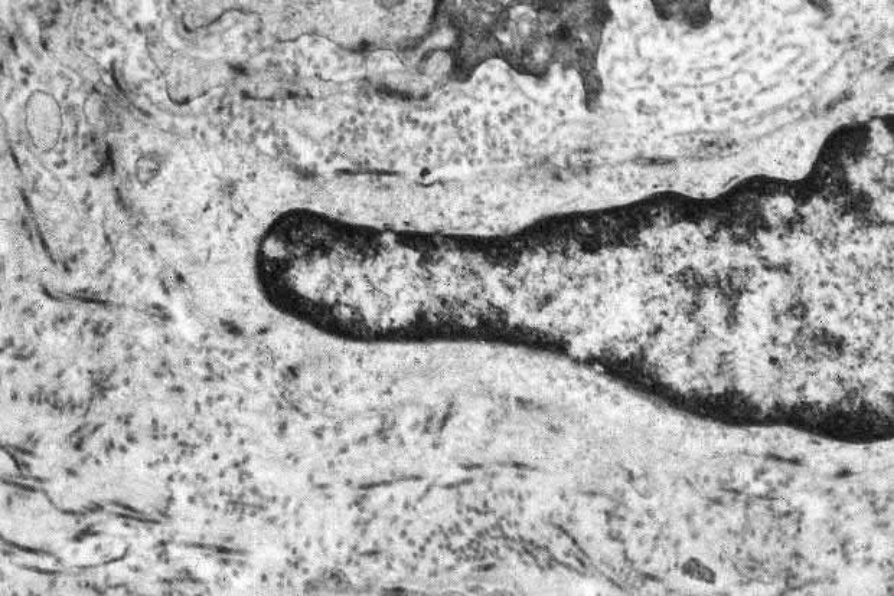
Fig. 13.115 Porphyria cutanea tarda: there is striking basement membrane reduplication surrounding this small dermal vessel.
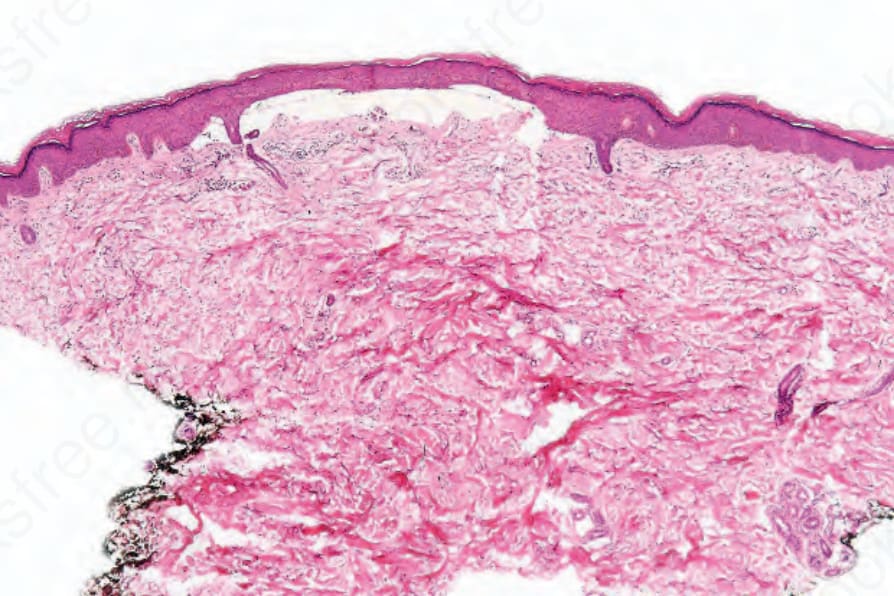
Fig. 13.116 Porphyria cutanea tarda: a bland subepidermal blister is present.
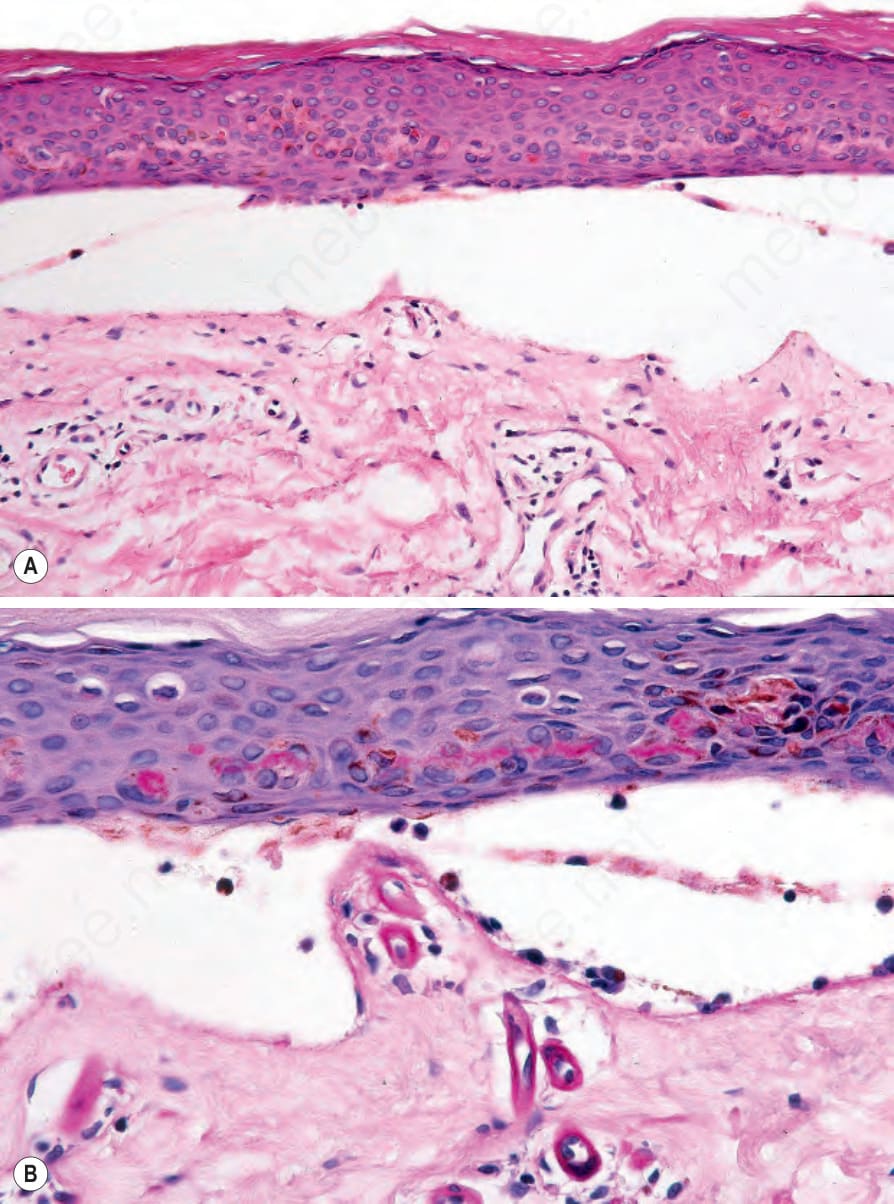
Fig. 13.117 Porphyria cutanea tarda: (A) the blister is cell free; (B) the superficial vessels are thickened (periodic acid–Schiff). Note the caterpillar bodies in the overlying epidermis.
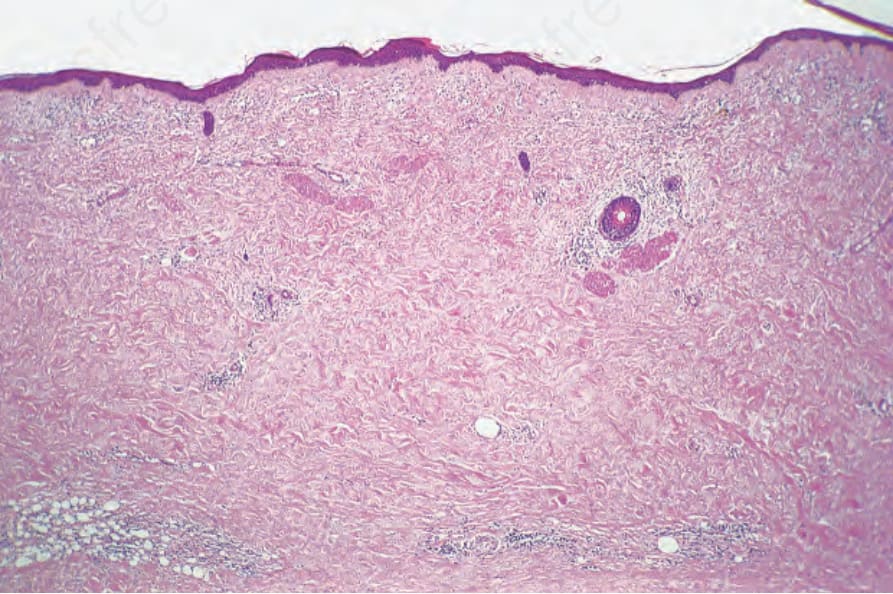
Fig. 13.118 Porphyria cutanea tarda: there is intense scarring of the entire dermis. The fat entrapment is reminiscent of scleroderma.
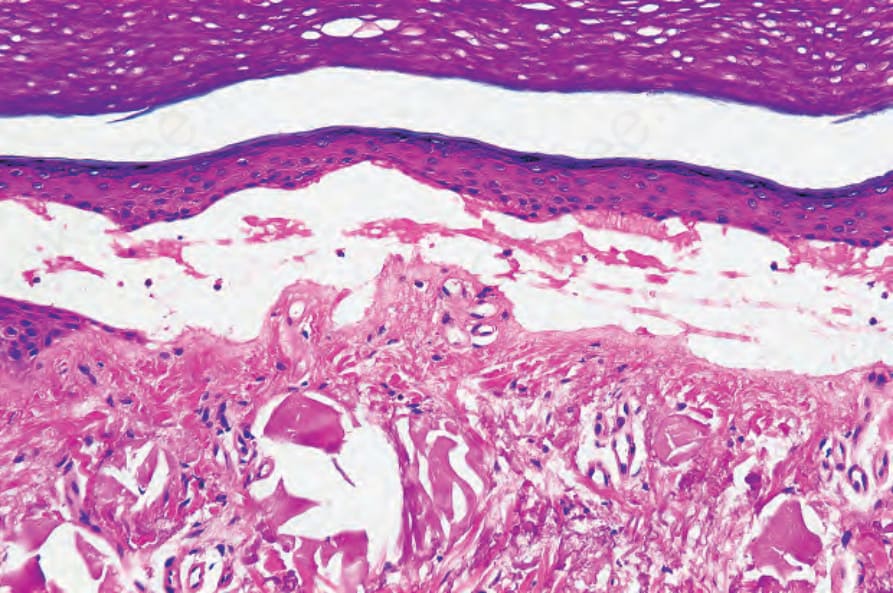
Fig. 13.119 Porphyria cutanea tarda: there is colloid milium-like solar elastosis deep to this blister.
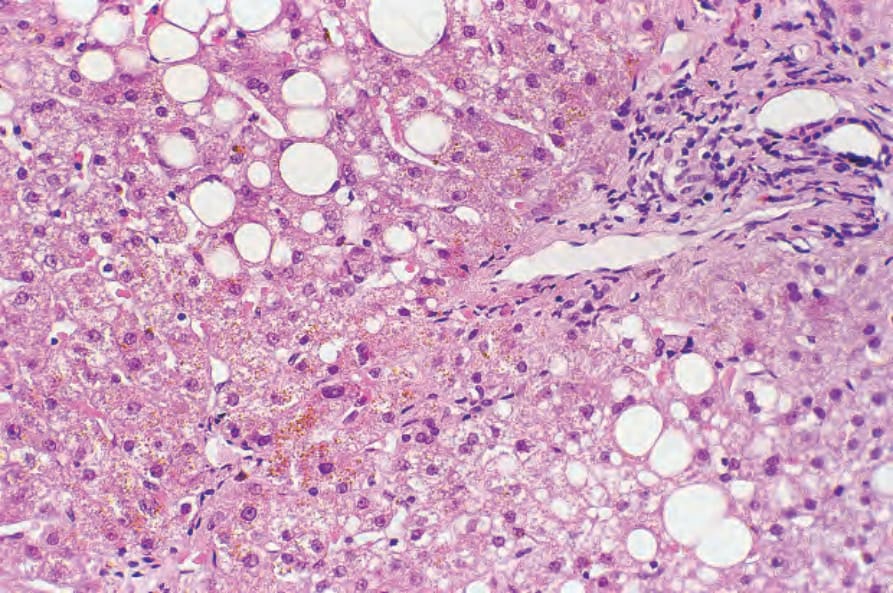
Fig. 13.120 Porphyria cutanea tarda: in addition to fatty change and mild chronic inflammation, brown uroporphyrin crystals are evident.
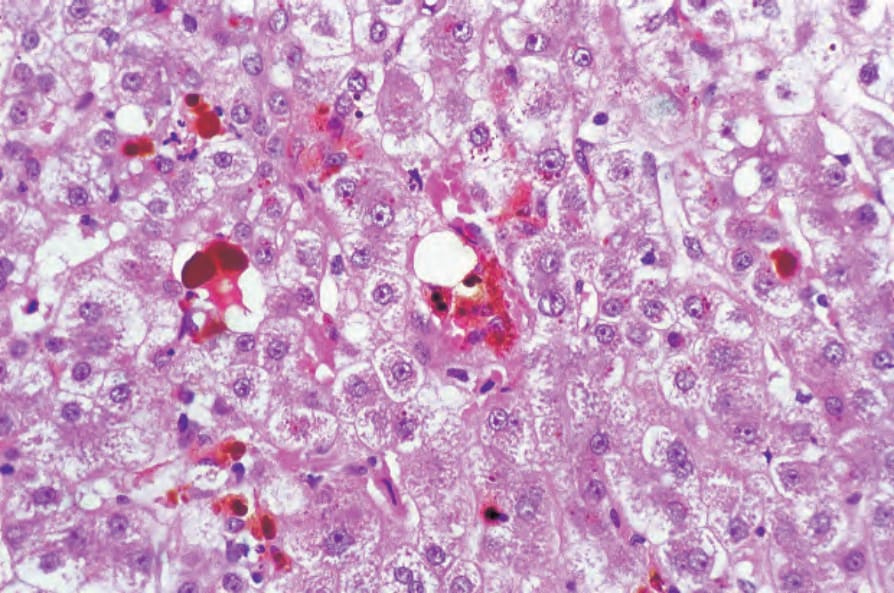
Fig. 13.121 Erythropoietic protoporphyria: the Kupffer cells contain abundant brown pigment. By courtesy of D.R. Davies, MD, St Thomas’ Hospital, London, UK.