Porphyria
Porphyria
The porphyrias constitute a heterogeneous group of conditions characterized by the excessive production of porphyrins or their precursors resulting from defects in the activity of the enzymes regulating heme synthesis (Fig. 13.91).1–12 Porphyrin synthesis occurs mainly in the erythropoietic
590 Degenerative and metabolic diseases
Condition Mode of inheritance Enzyme defect
Non-acute porphyrias producing cutaneous lesions Congenital erythropoietic porphyria
Site of metabolic expression Laboratory abnormality
Autosomal recessive Erythroid cells Elevated uroporphyrin Coproporphyrin in urine and feces
Porphyria cutanea tarda Urinary uroporphyrin: coproporphyrin = 3 : 1
inherited Autosomal dominant URO-D Hepatocytes Elevated urinary uroporphyrin
sporadic toxic
Acquired/sporadic Acquired
URO-D Urinary and stool isocoporphyrins
Erythropoietic protoporphyria
Autosomal dominant Ferrochelatase Erythroid cells and hepatocyte
Hepatoerythropoietic porphyria
Normal urine Elevated plasma, RBC and stool protoporphyrin Elevated fecal and RBC coproporphyrin
Autosomal recessive URO-D (severe) Erythroid cells and hepatocyte
Increased urine and stool URO Elevated stool coproporphyrin and isocoproporphyrin Elevated RBC protoporphyrin
Acute porphyrias (porphyrias producing abdominal, neurological, and psychiatric symptoms) Acute intermittent porphyria
Autosomal dominant Porphobilinogen deaminase Hepatocyte Stool and blood usually normal Elevated urinary ALA and PBG
ALA dehydratase deficiency
Autosomal recessive ALA dehydratase (porphobilinogen synthase)
? ALA alone elevated
Porphyrias producing abdominal, neurological, psychiatric, and cutaneous manifestations Variegate porphyria Autosomal dominant Protoporphyrinogen oxidase Hepatocyte Urine normal between attacks Increased stool protoporphyrins and coproporphyrin Increased urinary ALA and PBG during attacks
Hereditary coproporphyria Autosomal dominant Coproporphyrinogen oxidase Hepatocyte Increased stool and urine coproporphyrins
ALA, aminolevulinic acid; PBG, porphobilinogen; RBC, red blood cell; URO, uroporphyrinogen. Reproduced with permission from Young, J.W., Conte, E.T. (1991) International Journal of Dermatology, 30, 399–406.
system and the liver. Deficiency of a specific enzyme results in an accumulation of heme precursors due to stimulation of the rate-limiting enzyme aminolevulinic acid synthetase as a consequence of diminished heme concentration.10,13
intermittent porphyria, aminolevulinic acid (ALA) dehydratase deficiency, variegate porphyria, and hereditary coproporphyria). Of the eight major types of porphyria, six are associated with cutaneous disease (Table 13.4). The clinical and biochemical findings are very different in these six types of porphyria, although the cutaneous histology is similar in all.20–23 Type II porphyria cutanea tarda, hereditary coproporphyria, variegate porphyria, and erythropoietic protoporphyria are all inherited as autosomal dominants with incomplete penetrance. Fewer than 20% of affected individuals display symptoms and patients often deny a family history.2
Genetic mutations account for the enzyme deficiencies seen in the various types of porphyria. These mutations have all been delineated at a molecular level, are very heterogeneous, and often result in enzyme deficiencies that may remain silent throughout life.13 If a patient is homozygous for a specific mutation, however, symptoms usually develop even in early life.
Patients may present with acute porphyria (abdominal pain with neurological and/or psychiatric symptoms) often induced by drugs, fasting, alcohol or sex hormones.14–17 The enzyme defect leads to the accumulation in the skin of a photosensitizing porphyrin, which absorbs light predominantly in the 400–410 nm range. The energy absorbed may then be released to adjacent nucleic acids or proteins, either directly or indirectly by involving acceptor molecules, such as oxygen, and toxic changes causing damage to lysosomal and cellular membranes result.16,17 There is also some evidence to suggest that activation of the complement cascade may be involved in the phototoxic reaction mechanism.16,17 The cutaneous manifestations in acute attacks consist of prominent erythema in sun-exposed areas with a burning sensation. Subacute or chronic skin involvement consists of skin fragility, blister formation, and progressive scarring. Exceptional cases of a photosensitive bullous eruption associated with transient elevation of porphyrin levels have been described in neonates during phototherapy for treatment of hyperbilirubinemia due to hemolytic disease.18,19
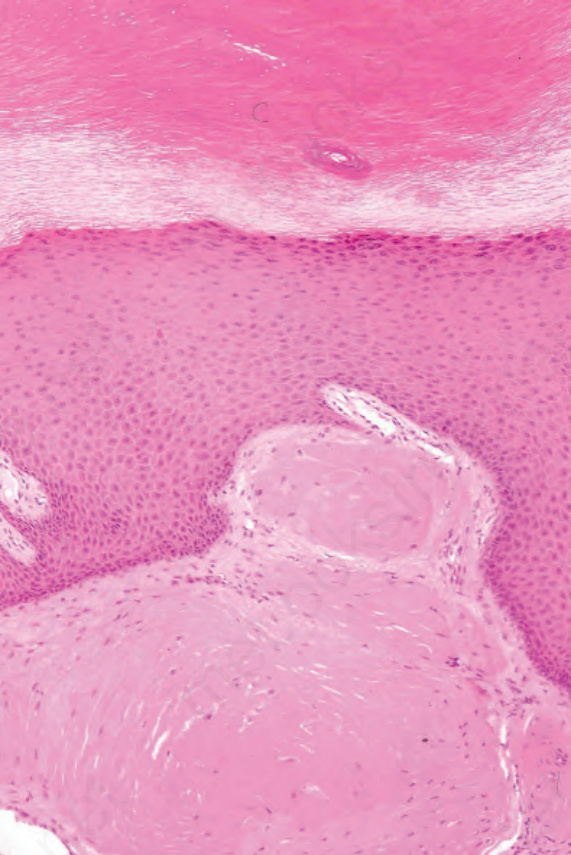
Fig. 13.89 Macroglobulinosis cutis: these are nodular deposits of eosinophilic material in the superficial dermis. By courtesy of A. Wang, MD, Brigham and Women’s Hospital, Boston, USA.
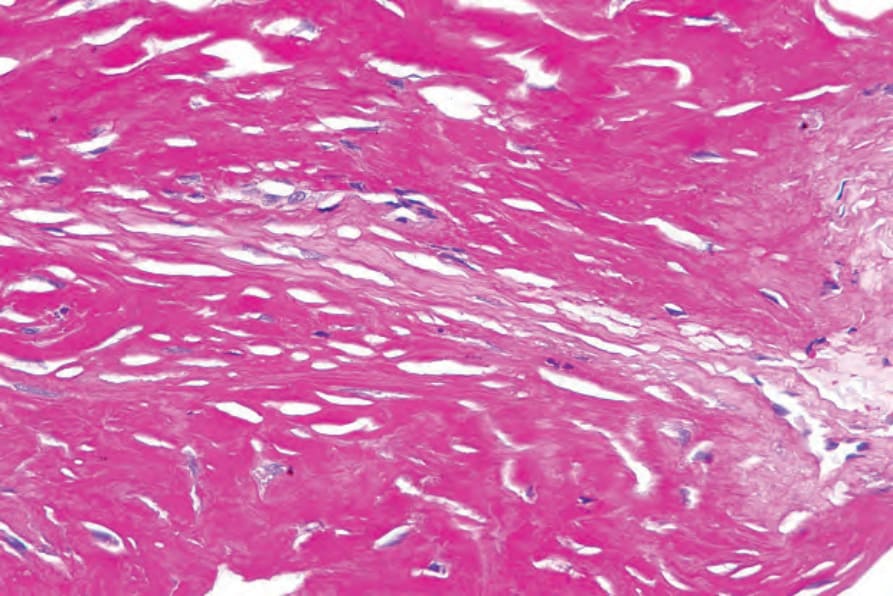
Fig. 13.90 Macroglobulinosis cutis: the material is strongly periodic acid–Schiff positive. By courtesy of A. Wang, MD, Brigham and Women’s Hospital, Boston, USA.
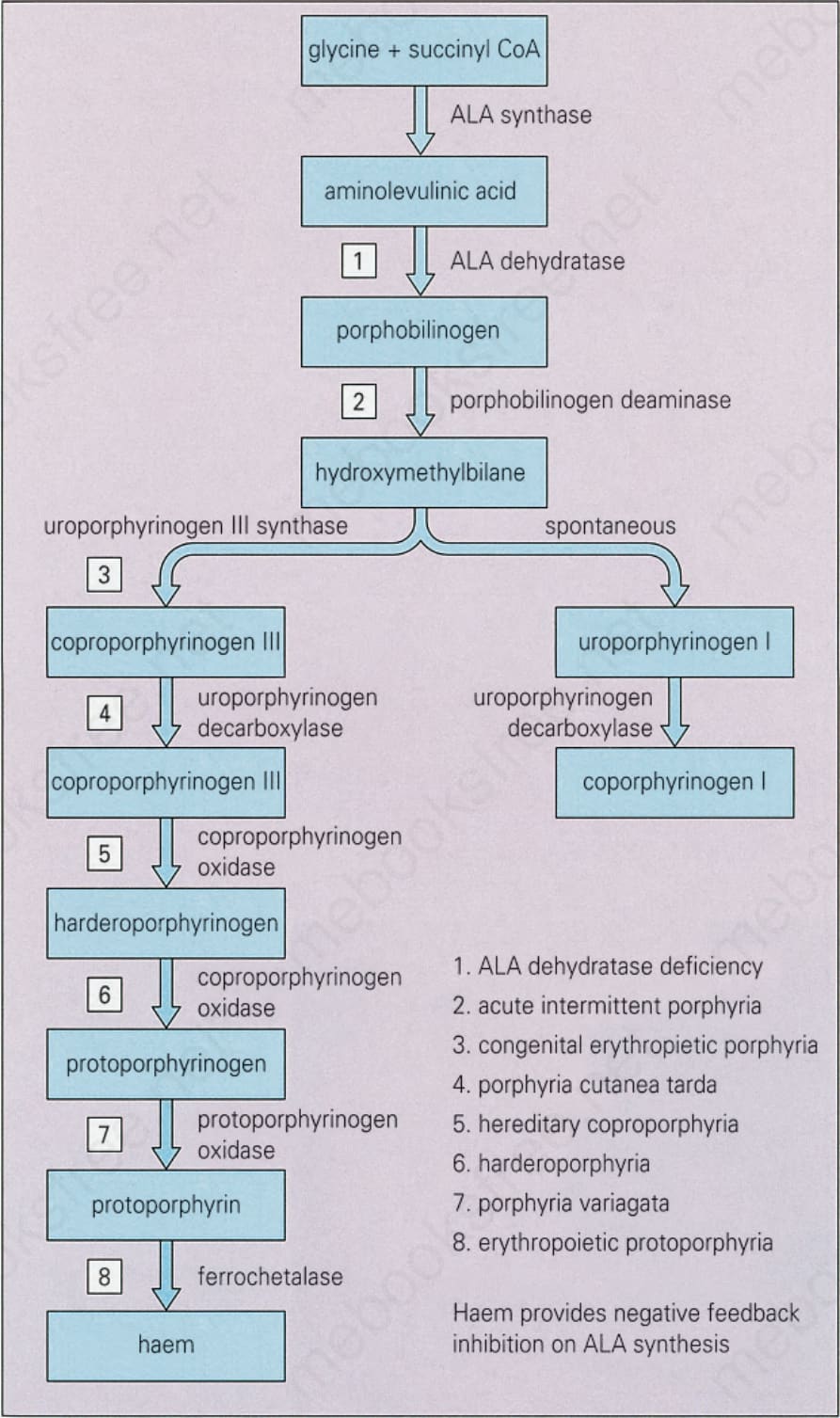
Fig. 13.91 Biochemistry of porphyria. Reproduced with permission from Young, J.W., and Conte, E.T. (1991) International Journal of Dermatology, 30, 399–406.
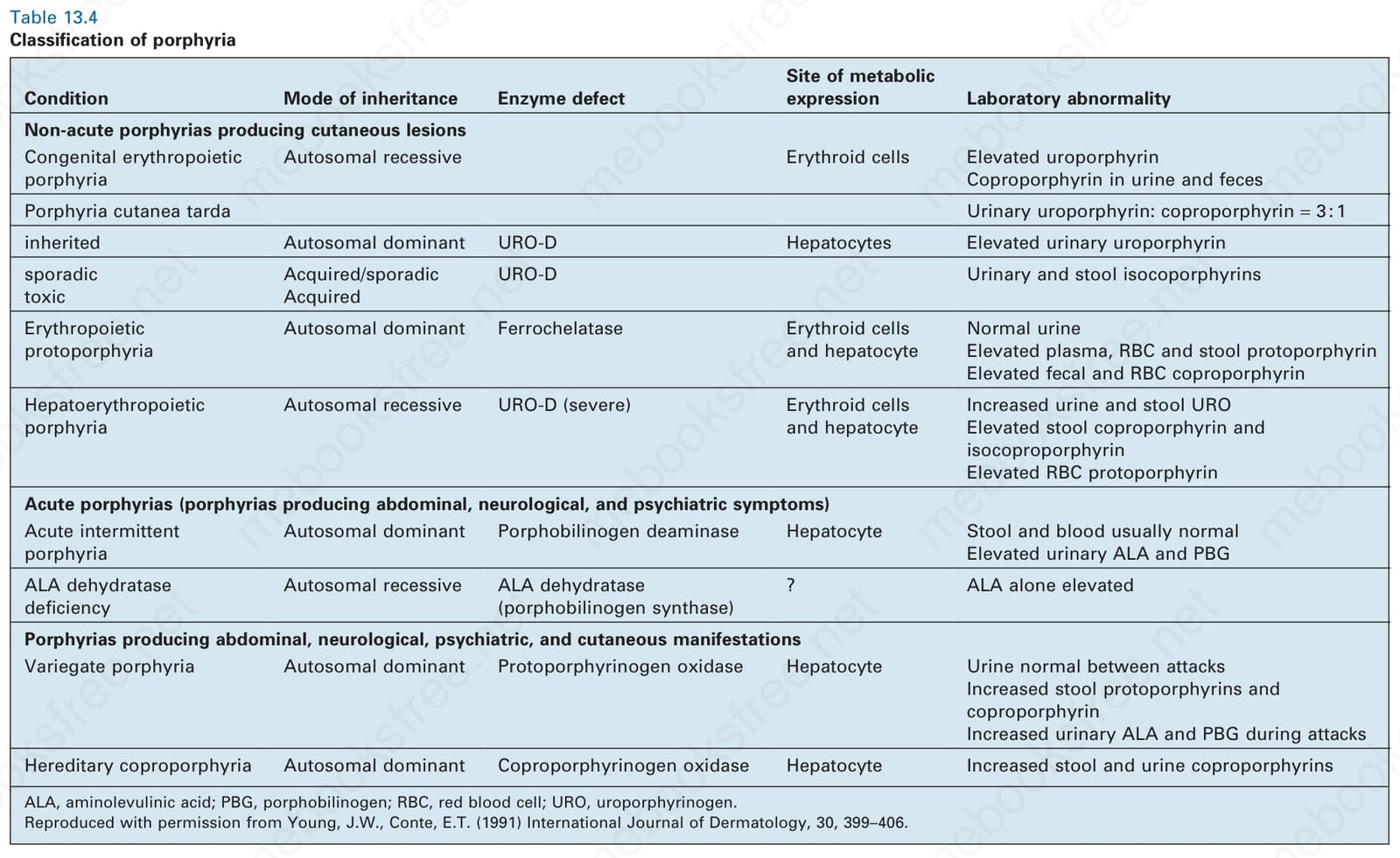
Table 13.4 Classification of porphyria