Sarcoidosis
Sarcoidosis
Clinical features Sarcoidosis (Gr. sarkos, flesh; eidos, form), so named because its histologic features were originally thought to resemble a sarcoma (Boeck), is a common systemic disease of unknown etiology. It is characterized and defined by the presence of noncaseating granulomata, usually (but not invariably) affecting multiple organ systems.1–11 Manifestations are variable. Patients may present with:
• an acute and usually self-limiting variant,
• a chronic form exclusively affecting the skin (up to between 20% and 40% of patients with cutaneous sarcoidosis do not have systemic involvement),
• a serious systemic chronic variant with widespread lesions, which affects multiple systems, is associated with high morbidity, and may occasionally be fatal. Sarcoidosis is more commonly encountered in industrialized countries and shows particularly high incidences in northern Europe (including the UK), the USA, and New Zealand, where as many as 20/100 000 of the population may be affected. It presents particularly in people in their third and fourth decades and shows a female predominance.12 In the USA, sarcoidosis is common among blacks, and there is a similar tendency in the UK (Figs 9.1 and 9.2). An epidemiological study of sarcoidosis in the Detroit, Michigan, area found that African-Americans living there had a 3.8 times greater risk of developing the disease compared with Caucasians.13 The disease is rare in Asians.14 First- and second-degree relatives of patients with sarcoidosis seem to have a significant risk of developing the disease compared to the normal population.15 The disease is rare in children, presents mainly in teenagers, and although the manifestations are usually similar to those seen in adults, infants may present with symptoms simulating juvenile rheumatoid arthritis (Fig. 9.3).16–18 Two forms of sarcoidosis have been identified in children. A variant affecting older children and occurring mainly during teenage years presents with a multisystemic disease similar to that seen in adults. Younger children under the age of 4 present with a cutaneous rash, arthritis, and uveitis.19 Infantile sarcoidosis should not be confused with Blau syndrome. This disease is inherited in an autosomal dominant fashion
and is characterized by sarcoidal granulomata in the skin, uveal tract, and joints but with no pulmonary involvement.20,21 Despite the similarities between both diseases, no genetic linkage has been identified.
Rarely, sarcoidosis presents in monozygotic twins.22 Coexistence with common variable immune deficiency is also a rare occurrence.23
Cutaneous lesions occur in 20–35% of patients with systemic sarcoidosis and may be classified into non-specific (erythema nodosum) and specific (granulomatous) subtypes.10 Cutaneous sarcoidal granulomata appear to be associated with a poorer prognosis and an increased incidence of pulmonary fibrosis and uveitis. Chronic facial lesions have been shown to be more commonly associated with involvement of the lungs, sinuses, and eyes.24 Erythema nodosum occurs quite commonly in sarcoidosis, with reported incidences varying from 11% to 31%.25 There is a significant female predominance (3 : 1). Interestingly, erythema nodosum appears to be relatively uncommon in both African-Americans and Caucasians in the USA. It presents as erythematous, tender, subcutaneous nodules, usually on the anterior tibial regions. Erythema nodosum may be associated with pyrexia, polyarthralgia (wrists, knees, and ankles), a very high erythrocyte sedimentation rate (ESR), and bilateral hilar lymphadenopathy (Lofgren syndrome). This acute form of sarcoidosis is associated with a good prognosis, with most patients experiencing resolution within 6 months of onset of symptoms.26–28 In one study, however, 16% of patients who presented with erythema nodosum developed chronic disease.27
A not uncommon mode of presentation is the development of a widespread, usually asymptomatic, maculopapular eruption. Individual lesions are erythematous or violaceous, 3–6 mm in diameter, and most commonly seen on the face (particularly in a periorbital distribution), the trunk, the extensor aspects of the extremities, and the neck (Figs 9.4 and 9.5). In this variant, the patient may also develop acute lymphadenopathy and uveitis, and a chest X-ray examination can reveal features of early respiratory involvement. Spontaneous resolution sometimes occurs. Occasionally, micropapular lesions are seen, particularly on the face and limbs (Fig. 9.6). Rarely, patients develop sheets of pinhead-sized lichenoid papules on the trunk and limbs. The onset is abrupt, and lesions may appear in crops. Some patients develop nodules and plaques, which may occur anywhere on the body, but most often affect the face, extremities, buttocks, and shoulders (Figs 9.7–9.11). Annular or serpiginous lesions are also encountered,
307 Sarcoidosis
and sometimes there is a prominent telangiectatic component (angiolupoid sarcoid) (Figs 9.12 and 9.13).10 Rarely, epidermal changes result in a psoriasiform or even ichthyosiform appearance.29 An exceptional case mimicking lipodermatosclerosis has been described.30 Chronic skin lesions are associated with pulmonary fibrosis, ocular, and bone involvement.
Most characteristic of sarcoidosis, however, is lupus pernio. This chronic violaceous plaque most often affects the nose, cheek, and ears, but lesions also sometimes affect the fingers and knees (Fig. 9.14). It is a particularly disfiguring variant, and resolution is especially complicated by marked scarring. Lupus pernio is often associated with lesions in the upper respiratory tract and can be followed by nasal obstruction and septal perforation. Patients also have severe pulmonary fibrosis, bone cysts, and ocular lesions. This variant has an insidious onset and is associated with a prolonged course and poor prognosis.16
Patients with sarcoidosis not uncommonly develop lesions in scar tissue31,32 and also in relation to trauma at the sites of desensitizing injections, tattoos, venipuncture, surgery, laser, cosmetic fillers, and BCG (Fig. 9.15).33–38 A single case attributed to copper-containing earrings has been described.39 Sarcoidal granulomata in association with foreign bodies do not necessarily imply a diagnosis of sarcoidosis. However, a small number of patients with
sarcoidal granulomata in association with silica and tattoo pigment may have systemic sarcoidosis or the latter may develop subsequently.40 Awareness of this problem is important as such cutaneous granulomata may be the first manifestation of the disease. Sarcoidosis has also been documented presenting in a tattoo in association with interferon-alpha (IFN-α) treatment for chronic hepatitis C.41 Interestingly, sarcoidosis has also been reported rarely in patients receiving interferon and ribavirin for chronic hepatitis C,42,43 and in rare patients on interferon-alpha or interferon-B therapy for melanoma.44,45 Sarcoidosis in patients with chronic hepatitis C may present concomitantly with the disease and unrelated to medication, or more often triggered by treatment, mainly ribavirin and interferon-alpha.46
308 Granulomatous, necrobiotic and perforating dermatoses
309 Sarcoidosis
310 Granulomatous, necrobiotic and perforating dermatoses
lupus pernio. Patients have interstitial fibrosis and eventual cor pulmonale, which may prove fatal.
Systemic vasculitis involving small- to large-caliber vessels has been found in some adults and children with sarcoidosis.77 This manifestation tends to be more common in African-American and Asian patients.77
Ocular lesions develop in about 20% of patients with sarcoidosis. Acute anterior uveitis is the most common manifestation; it is frequently bilateral and shows a predilection for females. It correlates with a benign outcome and erythema nodosum. Chronic uveitis also affects the anterior chamber and if untreated may progress to glaucoma and blindness. Other lesions include retinal vein perivasculitis, disc edema, and neovascularization. Conjunctival granulomata may be present in up to 30% of patients; therefore, biopsy can be a useful and relatively safe method of establishing the diagnosis. The lacrimal gland is also sometimes affected.
Neurological involvement occurs in 5–15% of patients with systemic sarcoidosis. Clinical manifestations include small fiber neuropathy, facial nerve palsy, Guillain-Barré syndrome, optic nerve disease, meningitis, seizure, and encephalopathy.6,78–80 In one study, neurosarcoidosis was the presenting symptom in 31% of patients.79 The combination of uveitis, facial nerve palsy, fever, and swelling of the parotid gland is known as uveoparotid fever (Heerfordt syndrome). This condition is often associated with central nervous system involvement. Hypothalamic and pituitary lesions are rare and may manifest as diabetes insipidus or panhypopituitarism.
Sarcoidosis has rarely been described in patients on Vemurafenib for metastatic melanoma.47 An association has also been occasionally reported in patients on infliximab, etanercept, adalimumab, ipilimumab, and natalizumab.48–50 Sarcoidal granulomas occurred in a patient on nivolumab for desmoplastic melanoma.51 The association of the disease with antitumor necrosis factor agents is paradoxical as these agents are used to treat the disease.
Peripheral lymphadenopathy develops in about 30% of patients. However, histologic examination of peripheral lymph nodes will reveal granulomata in about 75% of patients with sarcoidosis. Although splenomegaly is only present in 10–25% of patients, granulomata are present in about 50% of cases. Splenic disease is usually asymptomatic, but patients may have abdominal pain, hypersplenism, and, very rarely, splenic rupture. Splenic disease correlates positively with a high frequency of intrathoracic sarcoidosis. Liver function test abnormalities are quite common, and about 20% of patients have hepatomegaly; 60% of patients have hepatic granulomata on histologic examination.
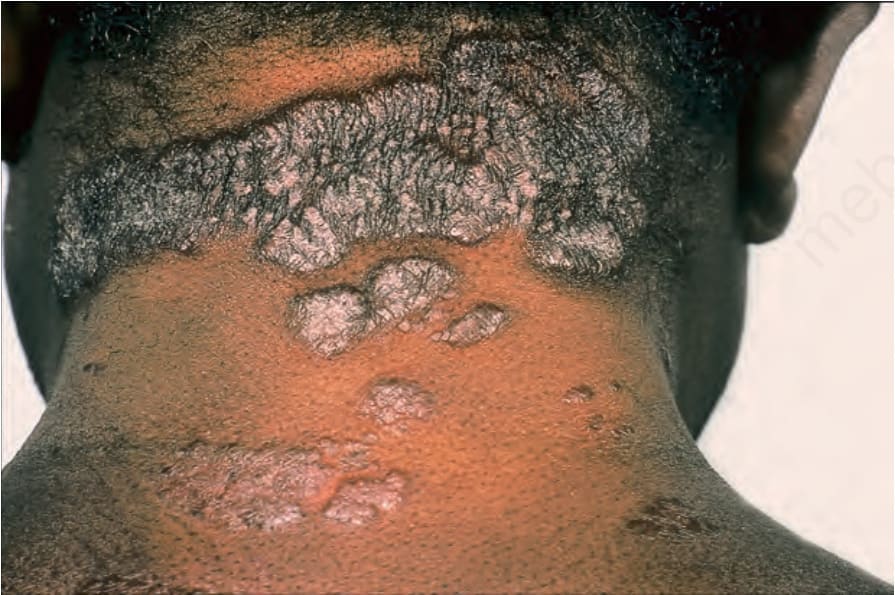
Fig. 9.1 Sarcoidosis: this patient presented with multiple plaques with raised margins on the neck. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
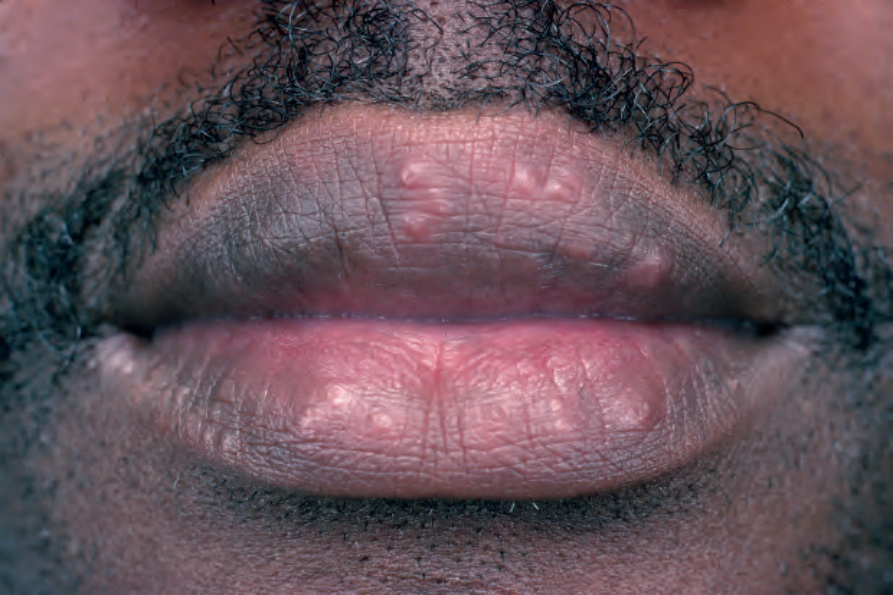
Fig. 9.2 Sarcoidosis: papules are present on both upper and lower lips. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
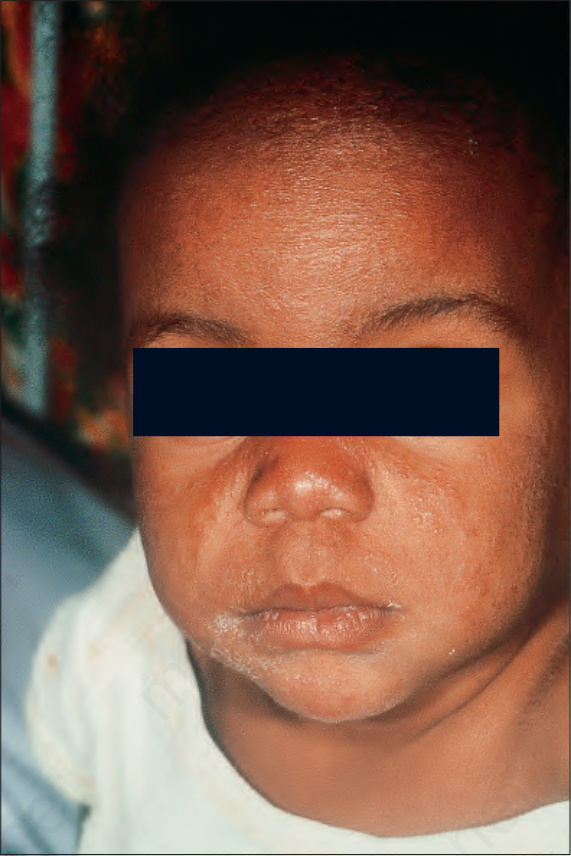
Fig. 9.3 Sarcoidosis: the condition is rare in children. There are widespread micropapules on this child’s face. By courtesy of C.T.C. Kennedy, MD, Bristol Royal Infirmary, Bristol, UK.
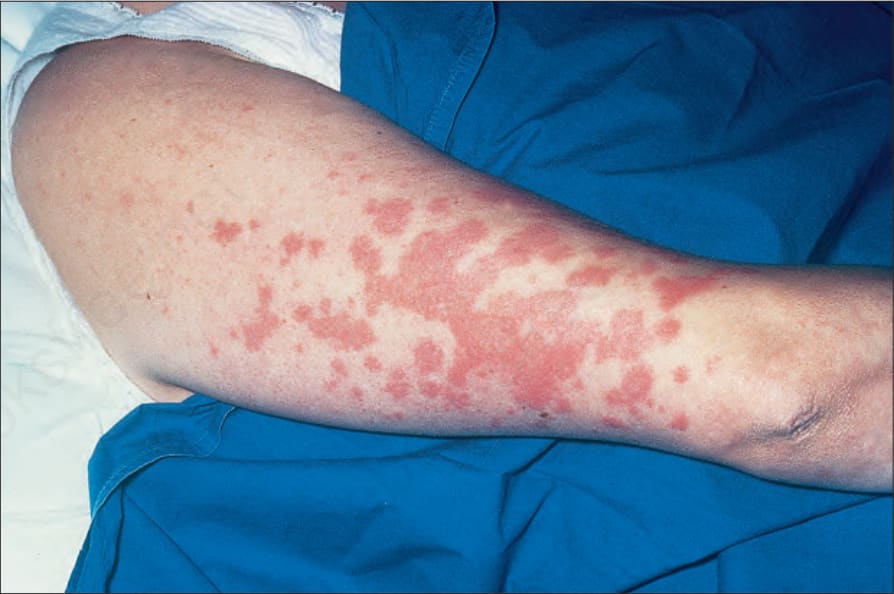
Fig. 9.4 Sarcoidosis: widespread erythematous plaques on the upper arm, some with an annular appearance. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
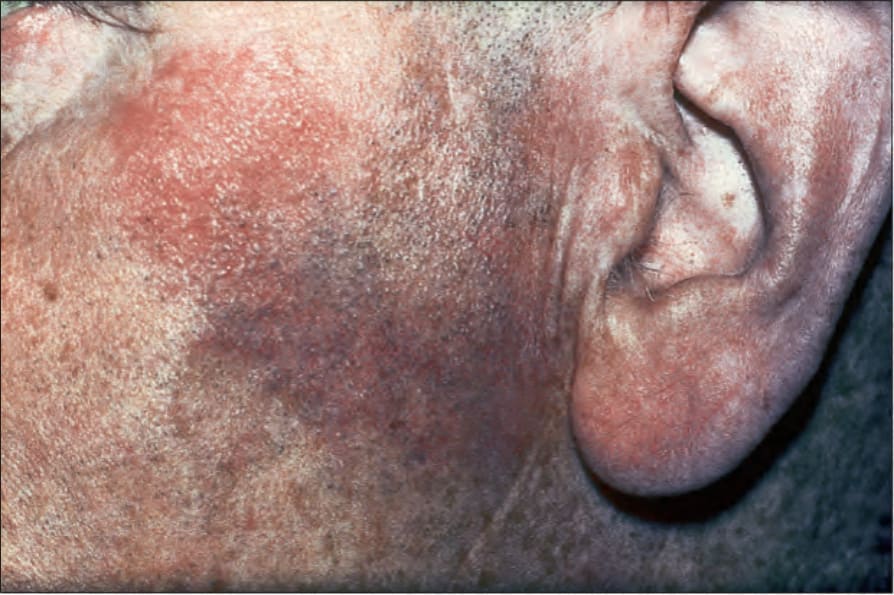
Fig. 9.5 Sarcoidosis: characteristic mauve plaque on the malar area with an infiltrative appearance. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
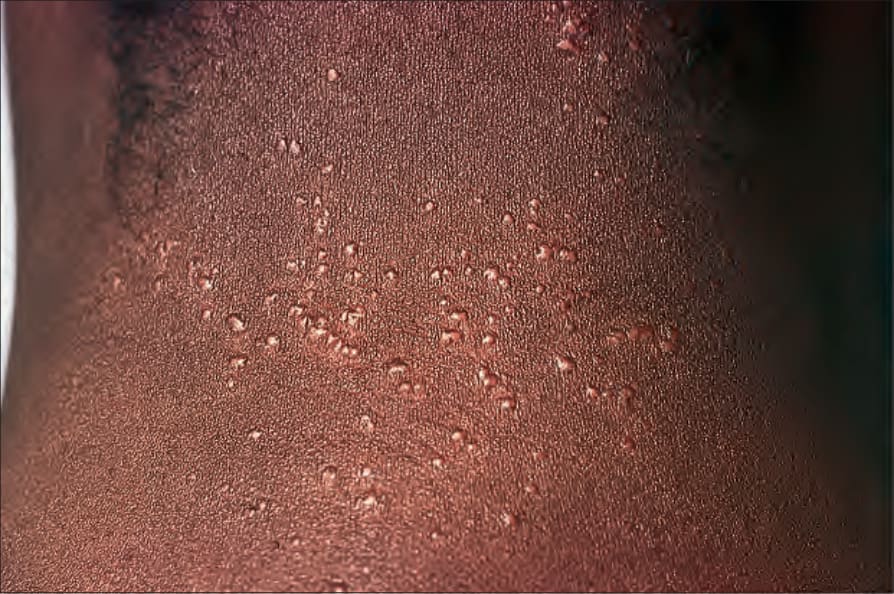
Fig. 9.6 Sarcoidosis: micropapular variant. Note the tiny lichenoid papules. By courtesy of the Institute of Dermatology, London, UK.
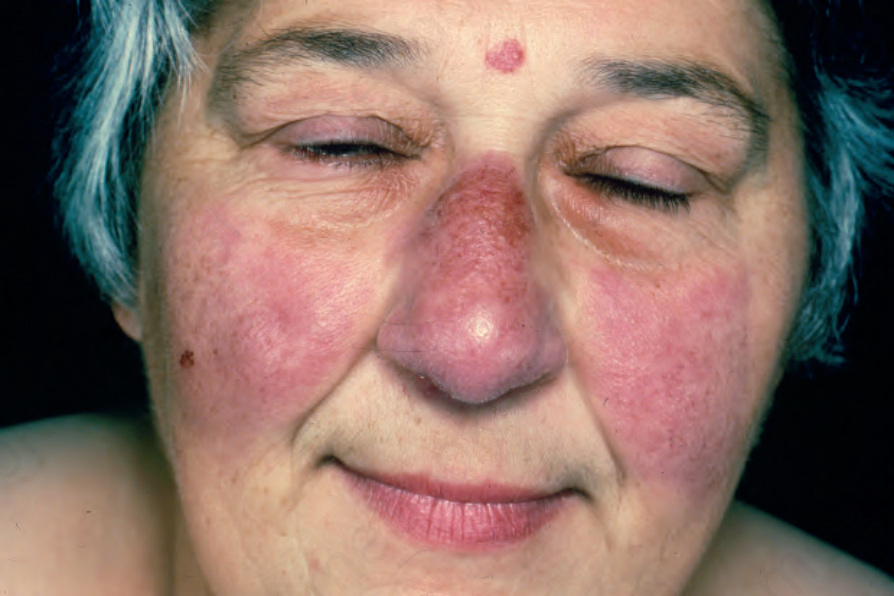
Fig. 9.7 Sarcoidosis: there is extensive facial involvement, a commonly affected site. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
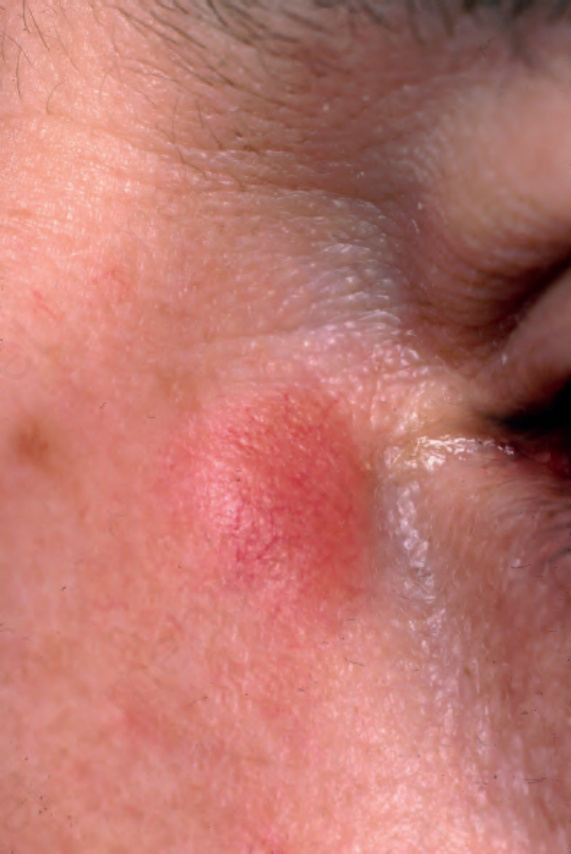
Fig. 9.8 Sarcoidosis: erythematous plaque adjacent to the eye. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
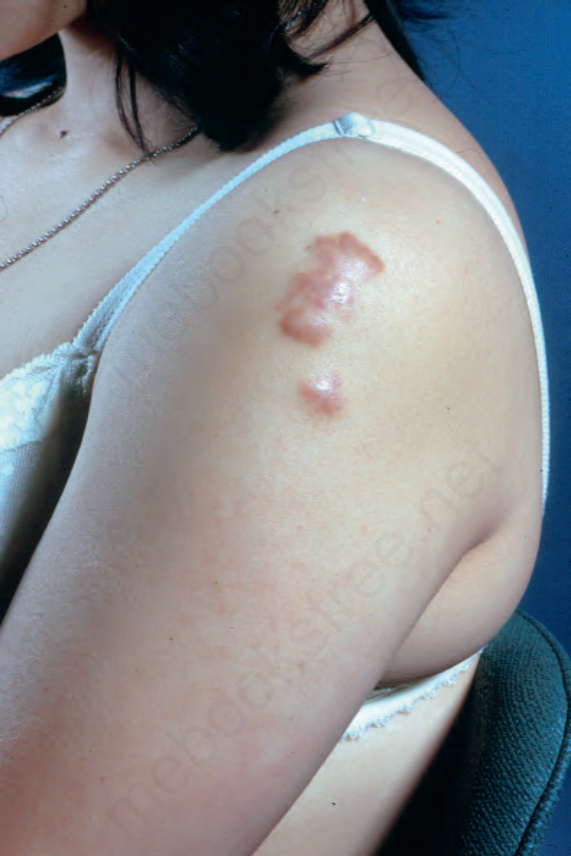
Fig. 9.9 Sarcoidosis: the extremities are commonly affected. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
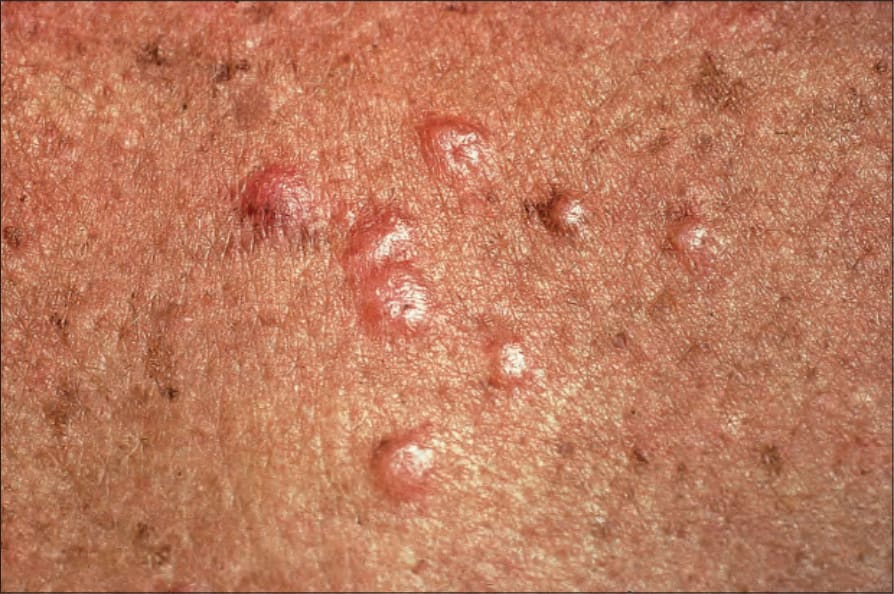
Fig. 9.10 Sarcoidosis: these grouped nodules are present on sun-damaged skin of the upper chest. By courtesy of the Institute of Dermatology, London, UK.
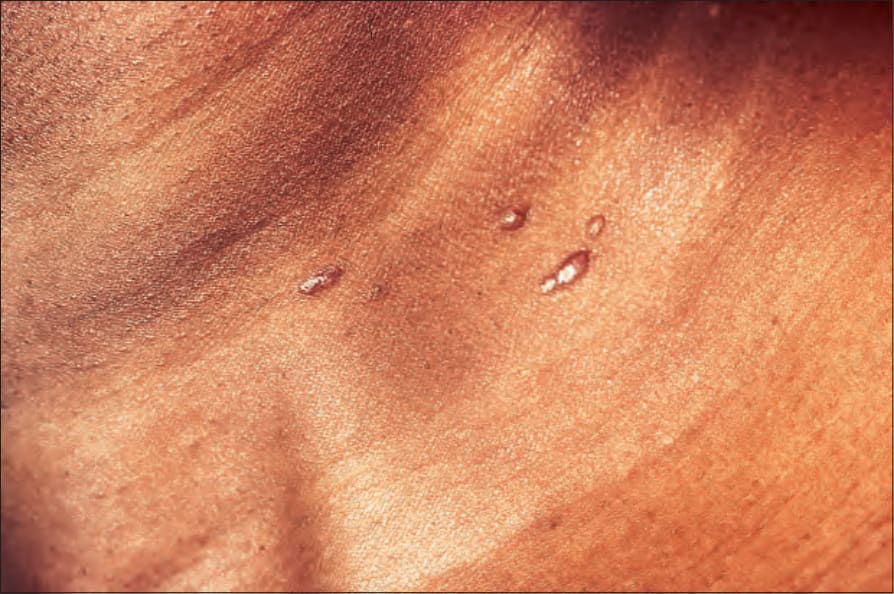
Fig. 9.11 Sarcoidosis: small nodules on the anterior aspect of the neck. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
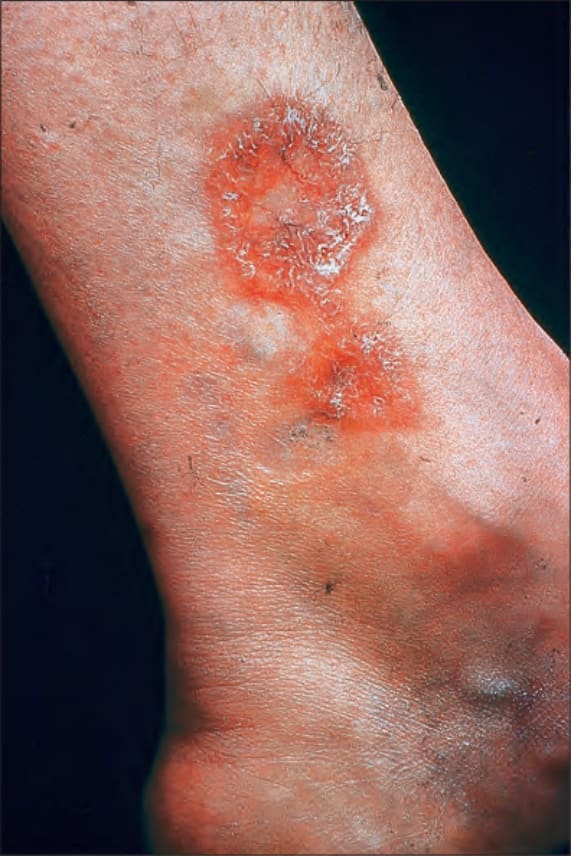
Fig. 9.12 Sarcoidosis: annular lesions on the ankle. By courtesy of the Institute of Dermatology, London, UK.
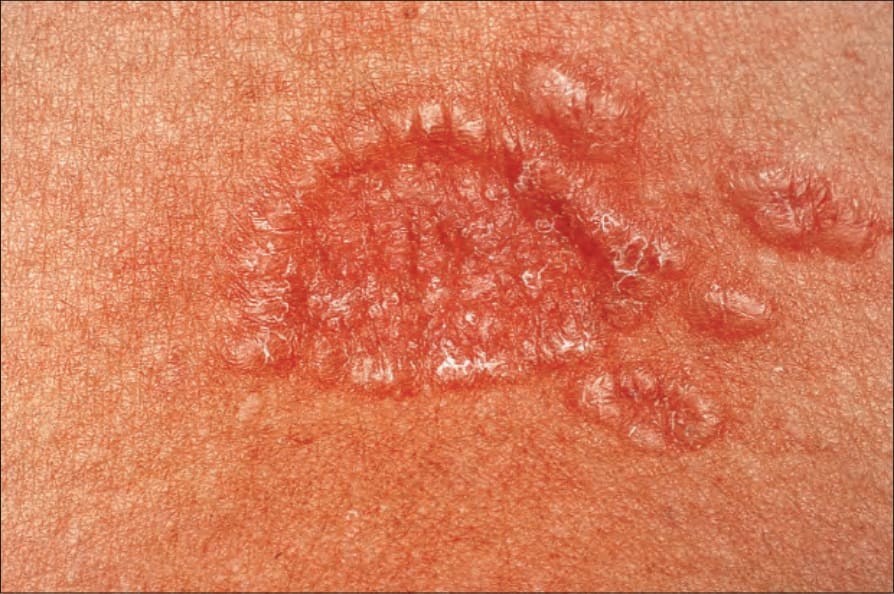
Fig. 9.13 Sarcoidosis: close-up view of an annular lesion. Note the beaded appearance. By courtesy of the Institute of Dermatology, London, UK.
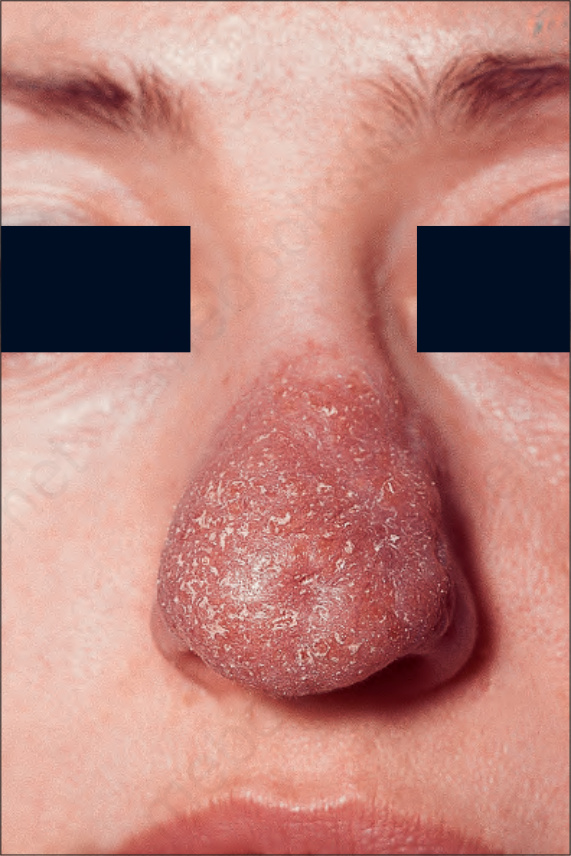
Fig. 9.14 Sarcoidosis: lupus pernio. The nose shows typical scaly violaceous swelling. By courtesy of the Institute of Dermatology, London, UK.

Fig. 9.15 Sarcoidosis: tattoo reaction. There are multiple dome-shaped nodules. By courtesy of the Institute of Dermatology, London, UK.
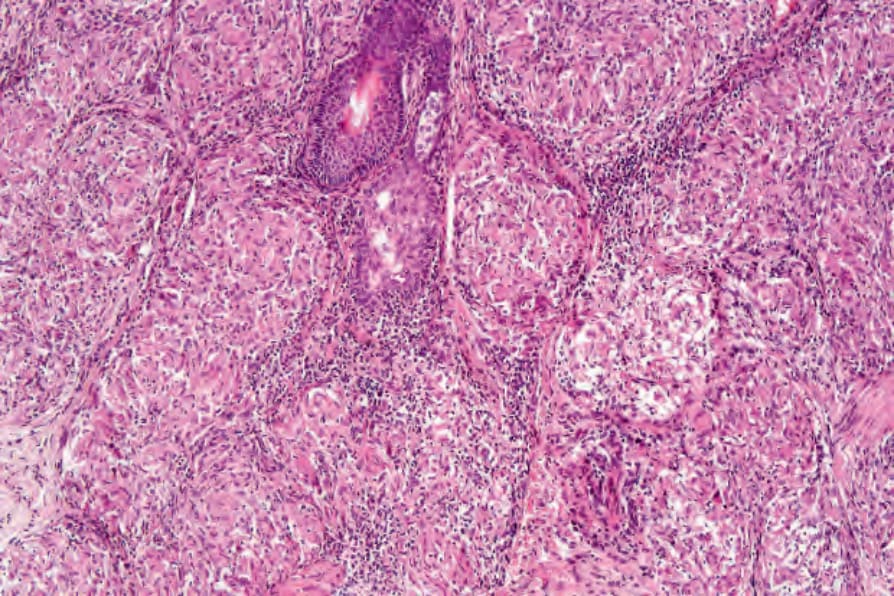
Fig. 9.18 Sarcoidosis: note the paucity of lymphocytes and absence of necrosis.
Cardiac lesions are uncommon, but are of particular importance due to the associated mortality.6 In an autopsy series, 50% of all deaths were due to cardiac disease.81 This same study found that the clinician often does not appreciate the presence of cardiac involvement – the antemortem diagnosis of cardiac lesions was made in only 29% of patients.81 Granulomata may occur at any site, but appear to show a predilection for the conduction system. Clinical manifestations include ventricular tachycardia, complete heart block, congestive cardiac failure, pericardial effusion, and myocardial infarction. Sarcoidal granulomata may affect small and large blood vessels, in particular the pulmonary vasculature.
Hypopigmented lesions may be seen in black patients.52 Unusual cutaneous manifestations include subcutaneous nodules, ichthyosiform lesions, erythroderma, scarring and nonscarring alopecia, lymphedema, nail dystrophy in the absence of underlying bone changes, verrucous lesions, generalized atrophy, leonine facies, palmar erythema, and leg ulcers with or without granulomatous vasculitis.53–67 Lesions of scarring alopecia may clinically mimic discoid lupus erythematosus.68 Subcutaneous nodules are rare and present as persistent, freely mobile, often painful lesions measuring 5–15 mm in diameter. It has been suggested that subcutaneous lesions are more often associated with systemic disease.69 In further studies however, subcutaneous involvement does not seem to be associated with increased incidence of systemic disease or worse prognosis.70,71 Oral and genital involvement is rare, but disease restricted to the vulva has been documented.72–74 Sarcoidosis has also been described presenting as a testicular mass.75 A very rare variant of seasonal photoinduced cutaneous sarcoidosis may occur.76
Muscle involvement is usually asymptomatic. Histologic examination of random muscle biopsies reveals granulomata in as many as 50% of patients. Rare features include asymptomatic palpable nodules and a polymyositis-like syndrome.
Although a rare complication, patients with sarcoidosis appear to be more prone to develop cryptococcosis than other infections.82
Radiologically demonstrable bone lesions occur in about 15% of patients. Early lesions consist of osteoporosis, cortical thinning, and mottled rarefaction. Established lesions are cystic and are sometimes associated with pathological fractures. The hands and feet are predominantly affected (Fig. 9.16). Destruction of the nasal bones can result from direct infiltration in patients with lupus pernio.
Ninety percent of patients with sarcoidosis have pulmonary involvement.28 Bilateral hilar lymphadenopathy is the most common intrathoracic manifestation of sarcoidosis and, together with pulmonary involvement, forms the most frequent lesion. Intrathoracic manifestations in sarcoidosis are classified into five subgroups: 6,10
• Stage 0: normal chest X-ray,
• Stage I: bilateral hilar and/or paratracheal lymphadenopathy with no pulmonary involvement,
• Stage II: lymphadenopathy with pulmonary infiltrates,
• Stage III: pulmonary infiltrates, but no lymphadenopathy,
• Stage IV: irreversible fibrosis and bullae, cysts, emphysema. Stage I disease is frequently associated with spontaneous resolution; progression to stage II disease is uncommon. Severe pulmonary involvement as seen in stage III patients correlates with deep chronic plaque lesions and
Hypercalcemia and, particularly, hypercalcuria are important complications of sarcoidosis. This is possibly due to increased intestinal absorption of calcium and abnormal production of 1,25-dihydroxyvitamin D.83 It is more often transitory, but in a small proportion of patients it is persistent and sometimes complicated by the development of renal failure due to nephrocalcinosis. Granulomata are found on histologic examination of the kidney in up to 40% of patients.
Laboratory investigations reveal a wide variety of abnormalities of the immune system (see below). Patients may demonstrate elevated levels of serum angiotensin-converting enzyme (ACE). Unfortunately, this finding is not specific for sarcoidosis, increased values also being found in patients with diabetes mellitus, alcoholic liver disease, and leprosy. It is sometimes
311 Sarcoidosis
cell wall deficient acid-fast bacteria (L forms) were cultured from the blood of 19 of 20 patients with sarcoidosis but not from controls.111 In summary, these mixed results between laboratories have not clarified the role of mycobacteria in the pathogenesis of sarcoidosis. It seems, however, that mycobacteria may be of etiological importance in at least a subset of cases.
Propionibacterium acnes DNA has also been identified in tissues of patients with sarcoidosis, including involved lymph nodes.112,113 The significance of this finding remains uncertain. Human herpesvirus 8 DNA has not been demonstrated in tissues of patients with sarcoidosis.113
The occasional association with known autoimmune diseases, such as progressive systemic sclerosis and systemic lupus erythematosus (SLE), has inevitably led to the proposal of an autoimmune pathogenesis. Although many familial cases have been reported in the literature, no consistent pattern of inheritance has emerged. The results of human leukocyte antigen (HLA) typing have shown associations with particular features of the disease; for example, HLA-A1 and HLA-B8 are associated with arthritis, HLA-A1 is also associated with uveitis, and HLA-B13 may be associated with a chronic refractory variant.10 Patients with HLA-DR17 have a better prognosis.114 One study has shown that patients with sarcoidosis have an increased frequency of a glutamine residue at position 69 of the B1 chain of the HLA-DPB molecule compared with a control population.115 This is particularly interesting since a similar polymorphism has been documented in patients with chronic beryllium disease, a disorder also characterized by granulomata and which shares some pathological features in common with sarcoidosis.
of value in monitoring the level of disease activity in patients known to have sarcoidosis. Patients may also display increased levels of serum and urinary lysozyme, serum beta-2-microglobulin, and collagenase.
Although sarcoidosis is associated with a high morbidity, the mortality rate is low, being of the order of 3–6%. Causes of death include cardiac involvement and respiratory or renal failure. The prognosis is better in females and appears to be improved in those with a positive purified protein derivative (PPD) skin test and normal serum immunoglobulin levels. The severity of disease is greater in blacks and Asians compared with Caucasians.84 Of interest, despite the very marked upset in immunological phenomena, patients do not seem to have an associated greatly increased risk of opportunistic infections except as a consequence of therapy (e.g., corticosteroids).
The association between sarcoidosis and a number of systemic diseases is probably coincidental. Sarcoidosis has been documented in association with vitiligo, pernicious anemia, autoimmune thyroiditis, Graves disease, chronic hepatitis, Addison disease, Sjögren syndrome, diabetes mellitus and ulcerative colitis, lymphoma, human immunodeficiency virus (HIV) infection, and primary biliary chirrosis.85–96 Interestingly, patients with acquired immunodeficiency syndrome (AIDS) usually develop manifestations of sarcoidosis after antiretroviral therapy is started. This phenomenon is the result of the immune restoration syndrome.93,97 Associations with cutaneous autoimmune disease include dermatitis herpetiformis and linear IgA disease.98,99 A single case of trachyonychia associated with sarcoidosis has been reported.100
Immunological investigations in patients with sarcoidosis have produced an immense wealth of data, which reveal that there is clearly an associated state of abnormal immunological hyperactivity. There are alterations of both cell-mediated and humoral immunity. Despite great efforts to clarify the immunobiology of sarcoidosis, particularly with regard to the precise antigens that may facilitate the disease, we still do not have a clear understanding of the disease process. Sarcoidosis, at least in part, appears to be due to a hyperactive T-helper cell proliferation with lymphokine production.11,116 Increased T-helper (Th1, Th2) cells are present in the alveolar lung parenchyma. Several studies have demonstrated selective activation of certain oligoclonal T-cell subsets.117–122 In one study, there was a correlation between the particular oligoclonal T-cell subsets and disease activity.117 Th1 lymphocytes (T cells expressing interleukin [IL]-2 and IFN-γ) preferentially accumulate in pulmonary parenchyma and the alveolar space compared with Th2 lymphocytes (T cells expressing IL-4 and IL-5).121 Compared with T cells in peripheral blood, T cells obtained by bronchoalveolar lavage show greater expression of IFN-γ and tumor necrosis factor-alpha (TNF-α).114 Of interest, patients with HLA-DR17 show a muted cytokine response, a finding that is perhaps related to the better prognosis observed in this subset of patients.115
Pathogenesis and histologic features The pathogenesis of sarcoidosis is poorly understood. The demonstration of familial clustering suggests hereditary susceptibility to sarcoidosis in at least a subset of patients.15,101
T lymphocytes, in turn, stimulate B cells. Abnormalities of humoral immunity include a non-specific polyclonal hypergammaglobulinemia and circulating immune complexes in acute forms of the disease, particularly in association with erythema nodosum.
The paramount puzzle in unraveling the pathogenesis of sarcoidosis is identifying the initial event(s) that lead to the disease. Despite our increasing knowledge of the immunobiology of sarcoidosis, we seem no closer to answering this key question. It seems clear, however, that the disease is the result of a complex interaction between many different external antigens, genetic predisposition, and the immune responses.
Despite intensive studies, the etiology and pathogenesis of sarcoidosis remains elusive.102,103 It is likely, however, that sarcoidosis represents a reaction pattern that may develop in a predisposed patient on exposure to one or more infective agents or other antigens.
The role of mycobacteria in the pathogenesis of sarcoidosis is a controversial topic. Attempts at detection of mycobacterial DNA by polymerase chain reaction (PCR) have produced conflicting results. While some authors have failed to detect mycobacterial DNA, others have identified DNA of various strains of tuberculous and nontuberculous mycobacteria.104–108 In one study, although amplified mycobacterial DNA was detected by PCR in 38% of sarcoidosis patients, mycobacterial DNA was also detected in tissue in 44% of control patients.109 Furthermore, most studies published in the literature fail to report more than 6% positivity for Mycobacterium tuberculosis DNA in patients with sarcoidosis.110 In another interesting study,
Histologically, sarcoidosis is characterized by a dense, noncaseating granulomatous infiltrate in the dermis (Figs 9.17 and 9.18), which sometimes extends into the subcutaneous fat. The granulomata are discrete and strikingly uniform in size and shape. They are composed of epithelioid histiocytes with abundant eosinophilic cytoplasm and oval or twisted vesicular nuclei often containing a small central nucleolus (Fig. 9.19). Variable numbers of Langhans giant cells are present, and sometimes a scattering of lymphocytes is seen at the peripheral margin of the granuloma (Fig. 9.20). Discrete small central foci of fibrinoid necrosis are sometimes present but caseation necrosis is rare (Fig. 9.21).122,123 Transepidermal elimination may be seen and seems more common than previously reported.124,125 The epidermis is usually normal although occasional cases display acanthosis and sometimes
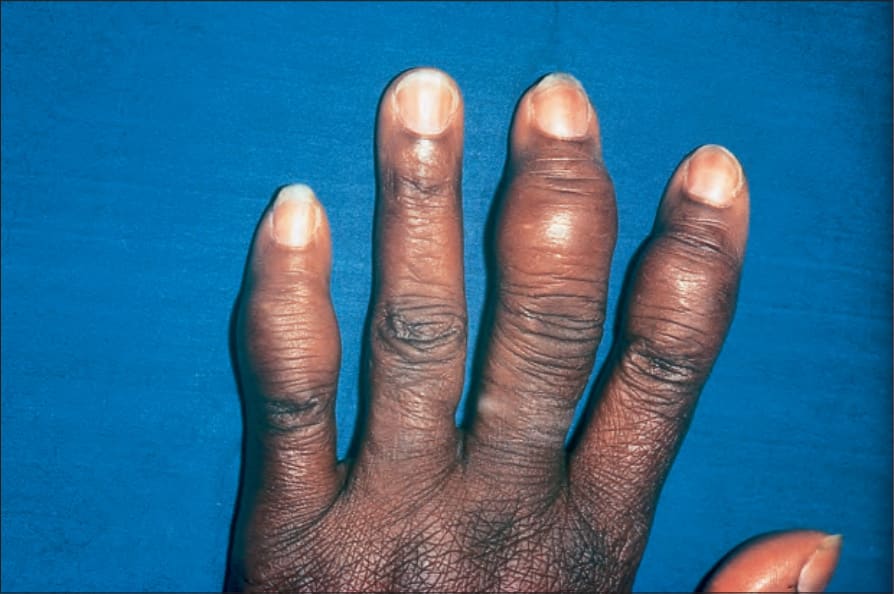
Fig. 9.16 Sarcoidosis: there is marked swelling of the distal interphalangeal joints. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
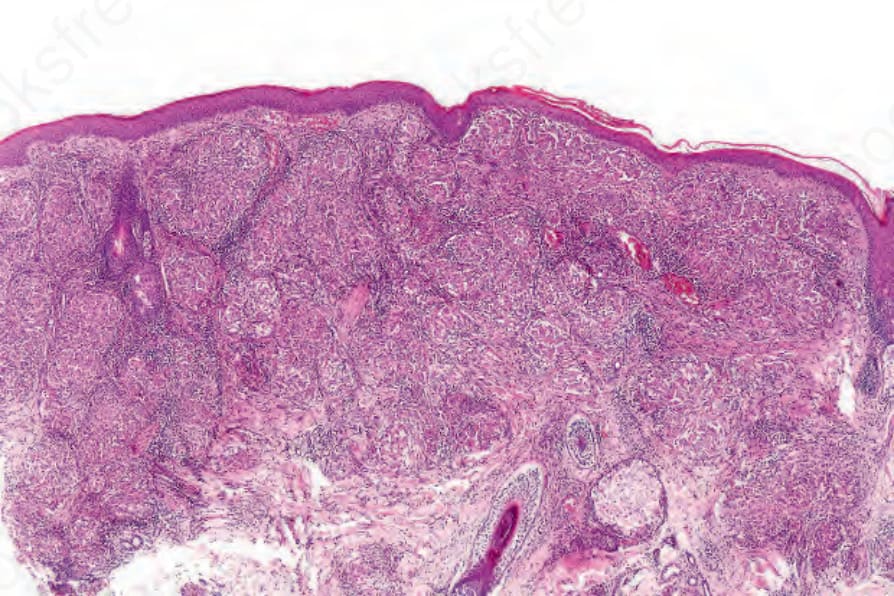
Fig. 9.17 Sarcoidosis: the dermis is replaced by uniform circumscribed nests of noncaseating granulomata.
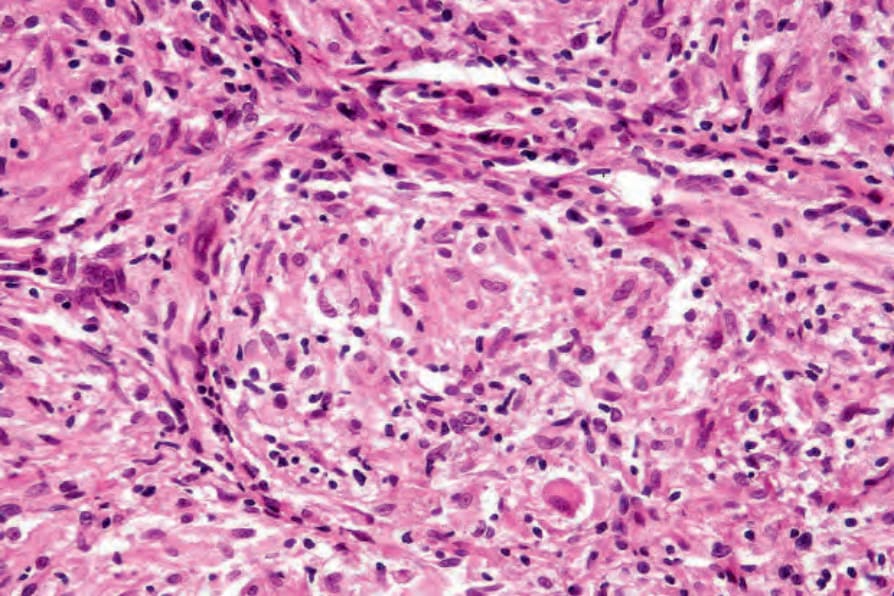
Fig. 9.19 Sarcoidosis: the epithelioid cells are composed of pink cytoplasm with a central oval or sometimes twisted vesicular nucleus containing a small basophilic nucleolus. The granuloma also contains lymphocytes and occasional fibroblasts.
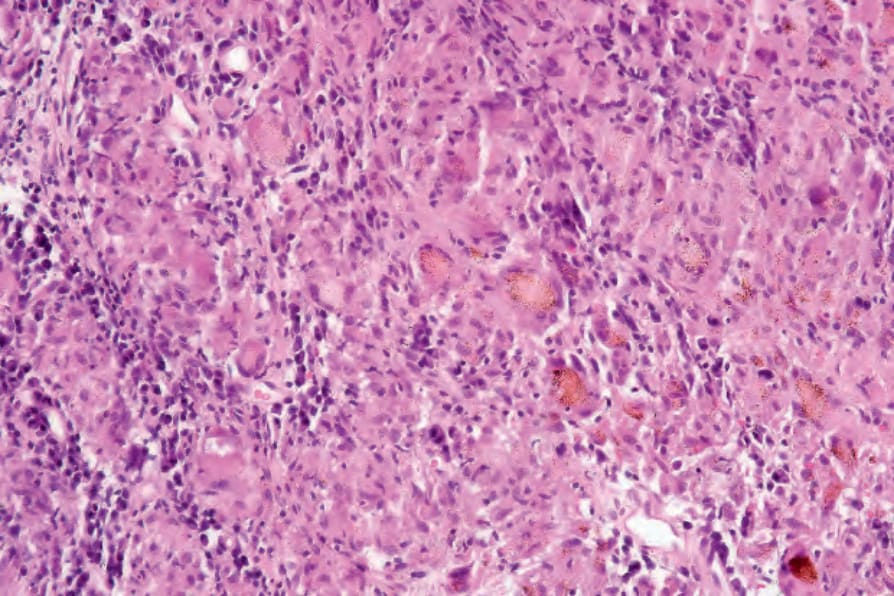
Fig. 9.20 Sarcoidosis: in this example, the granulomatous reaction is associated with abundant foreign material.
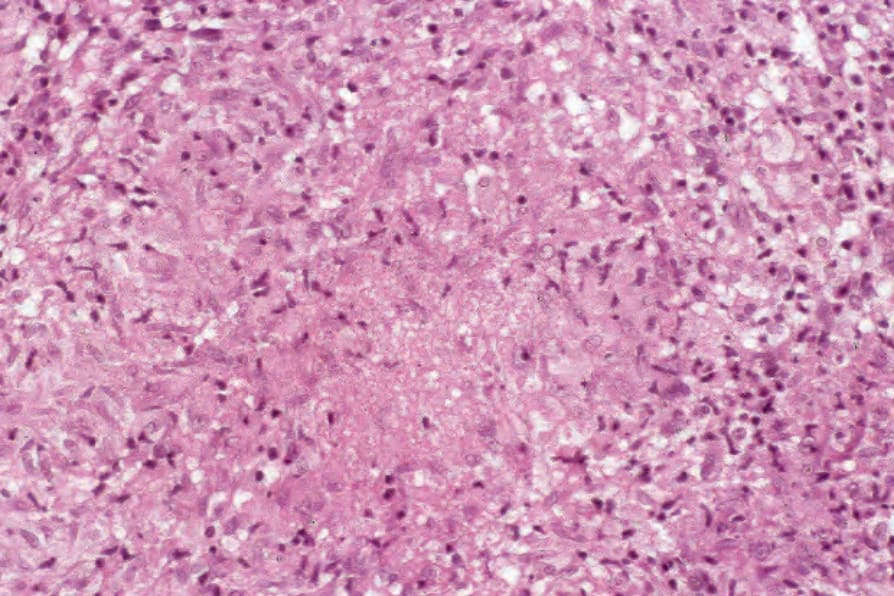
Fig. 9.21 Sarcoidosis: occasionally small foci of ‘fibrinoid’ necrosis may be seen in the center of the granuloma, but cellular detail is not lost.
312 Granulomatous, necrobiotic and perforating dermatoses
the granulomata are focally lichenoid. A predominantly lichenoid pattern may exceptionally be seen.126 A rare syringotropic variant with decreased sweating has been described.127 Exceptional cases of sarcoidosis may display histologic findings that focally overlap with granuloma annulare palisading neutrophilic and granulomatous dermatitis.128 Further histologic findings described include elastophagocytosis, perineural granulomas resembling leprosy, mucin deposition, and an infiltrate rich in plasma cells.78,129
In some cutaneous lesions, inclusion bodies are present, although much less frequently than in lymph nodes. The Schaumann body, a basophilic, laminated, rounded, conchoidal structure composed of calcium carbonate, calcium oxalate, phosphate, iron, and dolomite, is not specific for sarcoidosis and is seen in a number of other granulomatous conditions including tuberculosis and berylliosis (Fig. 9.22).129–131 The asteroid body is a small intracytoplasmic eosinophilic star-shaped structure; it is not specific for sarcoidosis, being seen also, for example, in tuberculosis, tuberculoid leprosy, berylliosis, and atypical facial necrobiosis. It is also commonly found in necrobiotic xanthogranuloma.132 Initial studies suggested that the asteroid body was composed of collagen, but more recent reports, using immunohistochemistry, suggest that it is a product of the microtubular system.133,134 The presence of foreign material in sarcoidal granulomata does not exclude
313 Granuloma annulare
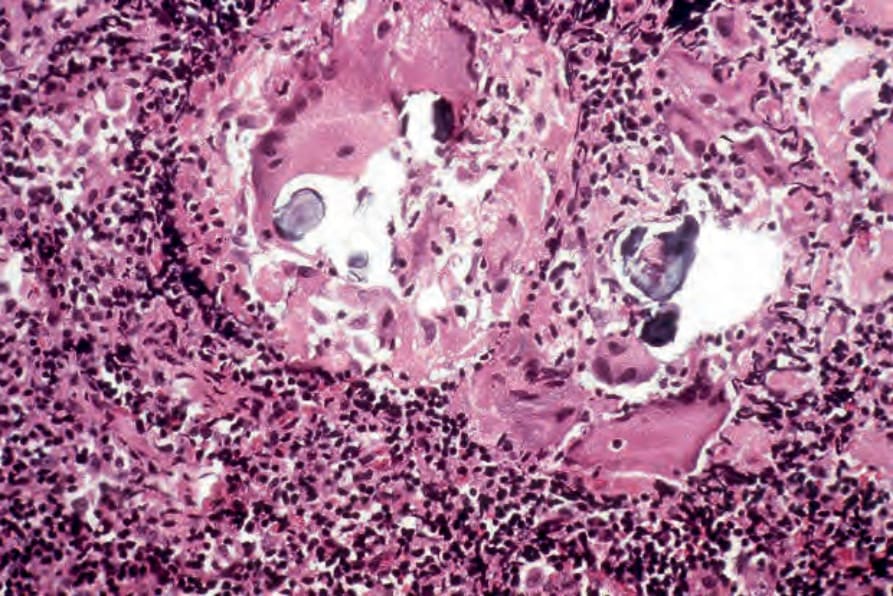
Fig. 9.22 Sarcoidosis: in this lymph node biopsy specimen, fragmented, laminated Schaumann bodies are seen. They are very rarely a feature of cutaneous sarcoidosis.