Urticarial vasculitis
Urticarial vasculitis
Clinical features Urticarial vasculitis is an uncommon condition characterized clinically by chronic urticaria and histologically by subtle leukocytoclastic venulitis.1–3 In some patients, urticarial vasculitis is associated with antibody–antigen complexes – a type III hypersensitivity reaction.4,5 In many patients, however, no underlying cause is discovered.
Patients may have, in addition to urticarial skin lesions, angioedema, arthralgia, gastrointestinal symptoms, and evidence of renal involvement.
The spectrum of illness ranges from mild symptoms to a serious systemic illness, for which treatment with corticosteroids is sometimes necessary.6
The disease shows a female predominance (2 : 1) and is most often seen in the third, fourth or fifth decade. It is rare in children.7 The cutaneous lesions are urticarial in appearance, but usually last 24–72 hours (Figs 8.58–8.60).8 Pruritus, a burning sensation, or pain, are common complaints. The frequency of attacks varies from daily to monthly. The skin lesions are edematous, raised, and erythematous, and are associated with nonblanchable purpura.
Systemic manifestations/associations include joint pain, stiffness and swelling, particularly of the hands, elbows, feet, ankles, and knees. Frank arthritis is extremely rare but may be associated with the development of valvular heart disease.9,10 Proteinuria and hematuria may be seen in some
303 Urticarial vasculitis
The erythrocyte sedimentation rate (ESR) is raised in many cases and in about 50% of patients there is hypocomplementemia. The presence of the latter correlates with systemic involvement.6,15 There may also be depression of the early classical pathway components C1q, C4, and C2. Patients with hypocomplementemic urticarial vasculitis have a high prevalence of autoantibodies to endothelial cells and antibodies against C1q are invariably present.16–19 The term ‘Schnitzler syndrome’ has been applied to patients with urticarial vasculitis and monoclonal IgM gammopathy.20–25 Hepatosplenomegaly, elevated ESR, elevated white blood cell count, fever, and joint pain are characteristic features and renal insufficiency has also been documented.21–23,26 An underlying lymphoproliferative disorder is present in a minor subset of patients.20
Importantly, urticarial vasculitis (especially the hypocomplementemic variant) is often associated with, or heralds the onset of, a variety of systemic diseases, including SLE, arthritis, interstitial lung disease, pericarditis, systemic sclerosis, mixed connective tissue disease, relapsing polychondritis, inflammatory myositis, hepatitis, inflammatory bowel disease, serum sickness, polyarteritis nodosa and Granulomatosis with polyangiitis, viral infections, Sjögren syndrome, cryoglobulinemia, polycythemia rubra vera, adverse reaction to drugs, and as a response to sunlight.6,15,27–45 In one study, more than 50% of patients had uveitis, scleritis, conjunctivitis, or episcleritis.6 It appears that patients with hypocomplementemia have more severe disease.27 Some authors have postulated that hypocomplementemic urticarial vasculitis represents a form of systemic lupus erythematosus.46 Others, however, have shown no significant difference in the association with lupus in patients with normocomplementemic compared with hypocomplementemic urticarial vasculitis.6 A diagnosis of urticarial vasculitis in any patient should initiate an evaluation for underlying disease. Fortunately, urticarial vasculitis usually has a benign outcome.6
patients. Many patients are hypocomplementemic.4,11 Rarely, renal biopsy reveals the features of focal or diffuse proliferative glomerulonephritis. Crescentic glomerulonephritis, and mesangial and membranous nephropathy have also been documented.6,12–14 Abdominal pain associated with nausea, vomiting, and diarrhea is a feature in some patients.
Urticarial vasculitis has been documented in association with malignancy.6,47–52 Given the rarity of this association, it may well be coincidental.
Histologic features In urticarial vasculitis, vascular damage is superimposed on a background of dermal edema and inflammation typical of urticaria. The vasculitis affects
304 Superficial and deep perivascular inflammatory dermatoses
the superficial vascular plexus. Extravasation of red blood cells is evidence of vascular damage. The vasculitis shows features of leukocytoclastic vasculitis except that the histologic features tend to be very subtle and are easily overlooked (Figs 8.61–8.63). Mild or focal fibrinoid changes associated with few neutrophils and sparse karyorrhexis are typical and eosinophils are almost always present. In our experience, the vasculitis is usually low grade in nature and often fibrinoid change is absent. Others have shown that endothelial necrosis is unusual.53 Nevertheless, more impressive necrotizing vasculitis may sometimes be encountered (Fig. 8.64). Urticarial vasculitis appears to represent a spectrum of disease ranging from urticaria with very mild vascular injury to frank necrotizing vasculitis.54–56
Differential diagnosis Clinical correlation is necessary to distinguish urticarial vasculitis from other forms of leukocytoclastic vasculitis. Although urticarial vasculitis is often associated with subtle, low-grade vascular injury, this pattern should not be relied upon in the distinction from other forms of vasculitis. In short, the pathologist’s role in diagnosis is to confirm the presence of vasculitis.
Tumor necrosis factor receptor-associated periodic syndrome (familiar Hibernian fever)
Clinical features Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) is a rare autosomal dominant condition with a predilection for individuals of Irish and Scottish descent.1–5 Other terms used to describe this disorder include familial Hibernian fever, benign autosomal dominant familial periodic fever, and autosomal dominant periodic fever with amyloidosis. Patients usually present in infancy or early childhood with prolonged episodes of fever accompanied by (in descending order of frequency) abdominal pain,
cutaneous manifestations, myalgia, headache, arthralgia, and pleuritic chest pain.3 Rare associations include central nervous system involvement due to a demyelinating disorder, IgA nephropathy, and small vessel vasculitis with concomitant relapsing panniculitis.6–8
Cutaneous manifestations include migratory asymptomatic, nonscaly erythematous macules and plaques, annular and serpiginous lesions measuring up to 28 cm in diameter.2,3 The trunk and extremities are predominantly affected with lesions presenting proximally and migrating distally.3 Patients may also develop conjunctivitis and periorbital edema.2 Amyloidosis is an occasional complication.9
305 Viral exanthemata
Cytomegalovirus Enterovirus (coxsakievirus, echovirus) Epstein-Barr virus Hepatitis B virus Human immunodeficiency virus Paramyxovirus Parvovirus Roseola (human herpesvirus-6) Rubella Rubeola (measles) Toga virus
Pathogenesis and histologic features TRAPS is due to a mutation of the TNFRSF1A gene which encodes the tumor necrosis factor receptor.3 The gene has been localized to chromosome 12p13.4 More than 60 mutations have been identified in the TNFRSF1A gene so far but the precise disease mechanism remains elusive.10
Histologically, the skin lesions are characterized by dermal edema and a superficial and deep perivascular and interstitial infiltrate of lymphocytes and a lesser number of macrophages.1–3 There is no evidence of vasculitis. Small numbers of neutrophils or plasma cells may occasionally be present.3
The infiltrate consists of CD3+ T lymphocytes and CD68+ macrophages. CD4+ T-helper and CD8+ T-suppressor forms are both represented. CD20+ B lymphocytes are not present.
accompanied by spongiosis. Other manifestations include eosinophilic spongiosis, pustules, bullous pemphigoid-like subepidermal blistering, and eosinophilic panniculitis.
Direct immunofluorescence may disclose perivascular deposits of C3 and IgM. Indirect immunofluorescence studies are negative.
The lymphocytes are of the CD3+ T-cell phenotype with a predominance of CD4+ T-helper cells over CD8+ T-suppressor cells. B cells are absent.
Eosinophilic, polymorphic, and pruritic eruption associated with radiotherapy
Clinical features Rueda and coworkers documented a polymorphic cutaneous eruption which develops in females undergoing radiotherapy for cancer (eosinophilic, polymorphic, and pruritic eruption associated with radiotherapy, EPPER).1–6 Although a wide spectrum of tumors may be present, cervical squamous carcinoma and breast carcinoma are by far the most common. The cutaneous lesions are intensely pruritic and include excoriations, erythematous papules, vesicles, blisters, pustules, and panniculitis-like lesions.1–8 The extremities, particularly the legs, are predominantly affected.
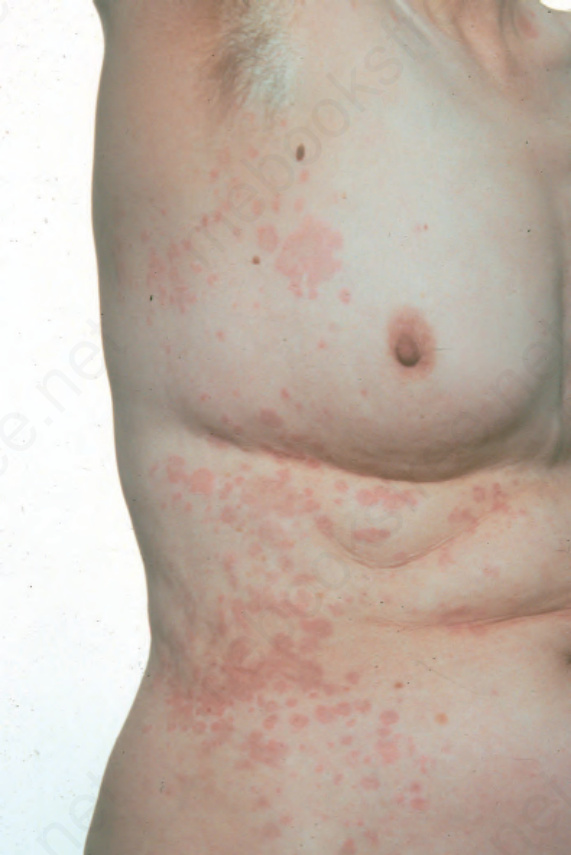
Fig. 8.58 Urticarial vasculitis: note the urticaria with a livid hue. By courtesy of J. Newton, MD, St Thomas’ Hospital, London, UK.
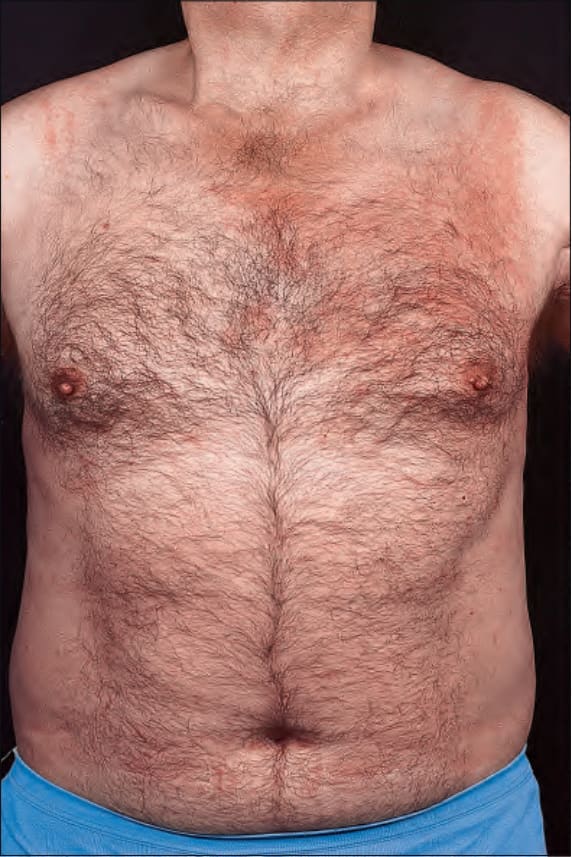
Fig. 8.59 Urticarial vasculitis: in this patient, there is an extensive urticarial plaque. By courtesy of J. Newton, MD, St Thomas’ Hospital, London, UK.
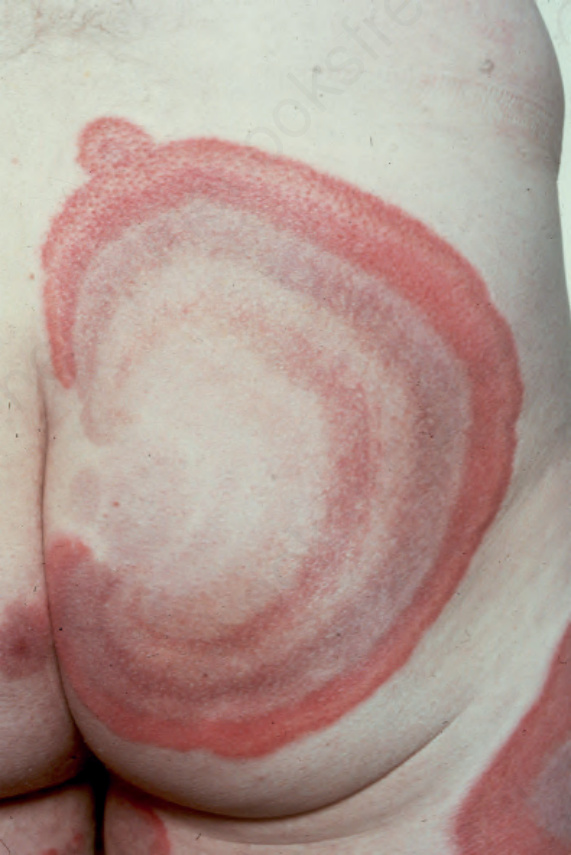
Fig. 8.60 Urticarial vasculitis: note the bizarre annular purpuric urticarial plaque. By courtesy of J. Newton, MD, St Thomas’ Hospital, London, UK.
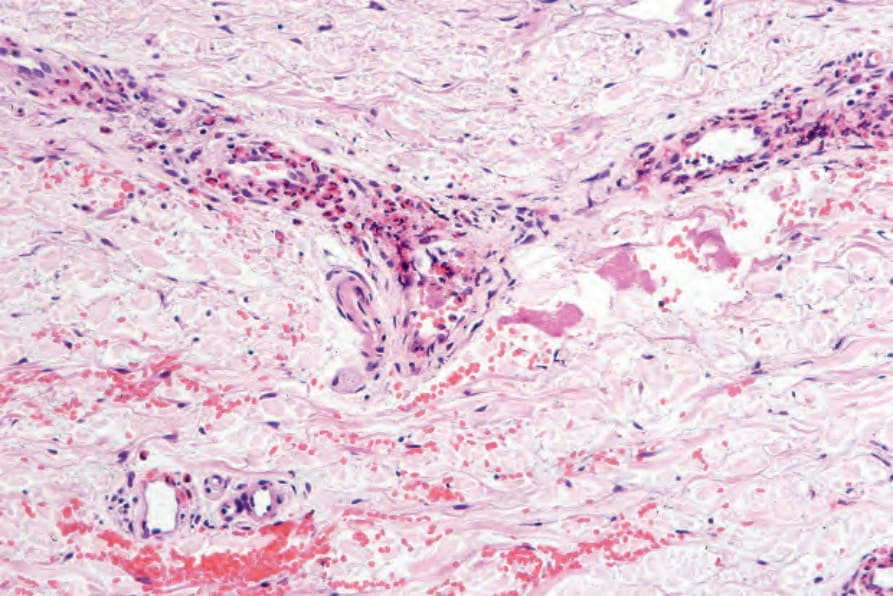
Fig. 8.61 Urticarial vasculitis: in this example of an early lesion, there is a conspicuous perivascular eosinophil infiltrate. There is no evidence of vessel wall damage.
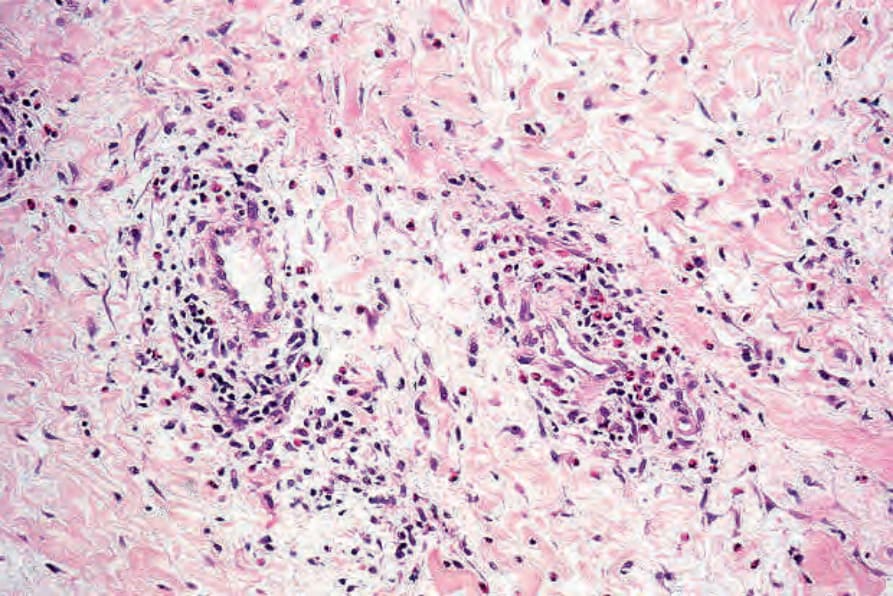
Fig. 8.62 Urticarial vasculitis: in this field, there is marked edema accompanied by a mixed lymphocytic and eosinophil infiltrate.
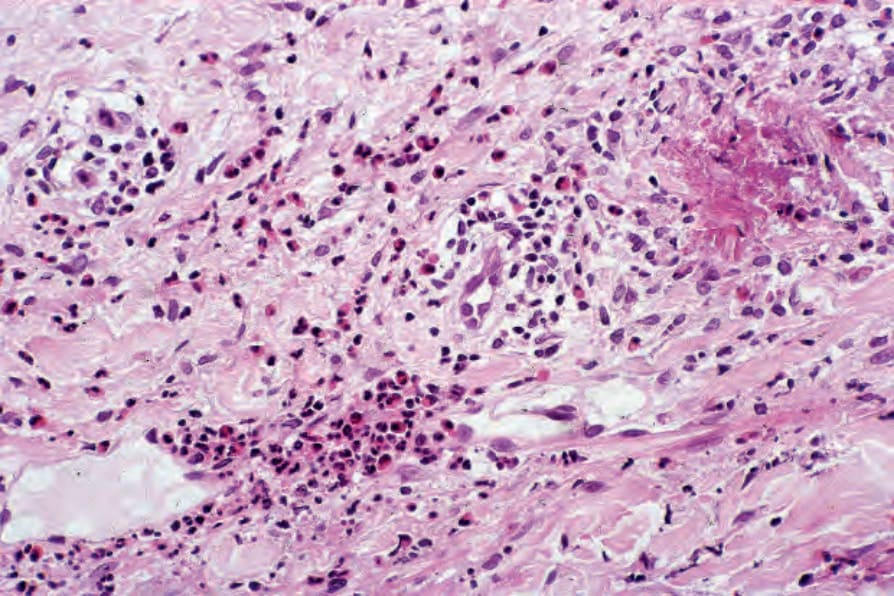
Fig. 8.63 Urticarial vasculitis: there is a heavy lymphocytic and eosinophil infiltrate. In this example, there are conspicuous flame figures.
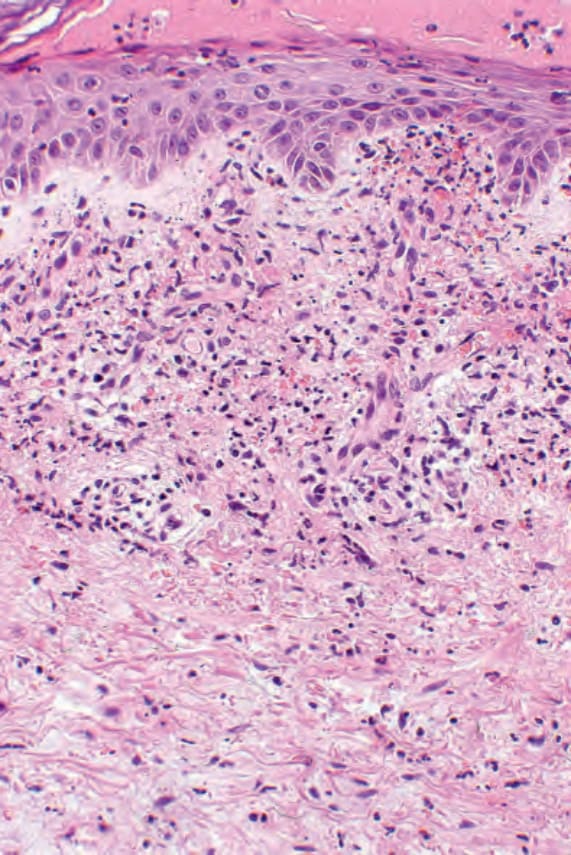
Fig. 8.64 Urticarial vasculitis: this biopsy of a purpuric lesion shows features of florid vasculitis.

Table 8.1 Viruses associated with cutaneous eruptions