Bullous pemphigoid
Bullous pemphigoid
Clinical features Bullous pemphigoid is not a single disease entity. Rather, there are many subtypes, which have been classified into primary cutaneous and mucosal variants and into generalized and localized forms (Fig. 4.40).1–4 Bullous pemphigoid (BP) is the most frequently encountered autoimmune bullous dermatosis with an annual incidence of 6.6 new cases per 1 million of the population.5,6
Generalized cutaneous pemphigoid Any age group may be affected, but the generalized variant demonstrates a predilection for the later years of life, showing a maximum incidence in the seventh decade and over. Rarely, however, children and even infants may be affected.7,8 The disease is associated with a worldwide distribution and shows no racial propensity. There are no significant human leukocyte antigen (HLA) associations and the sex incidence is approximately equal.
Prodromal events are numerous and include erythematous, urticarial and, rarely, eczematous phases.9,10 Erythroderma, either preceding the bullous phase or occurring simultaneously, is a very rare manifestation (erythrodermic pemphigoid).11,12 Similarly, patients may present with a history of generalized pruritus in the absence of visible skin lesions (pruritic pemphigoid). In such circumstances, immunofluorescence investigations are essential to establish the correct diagnosis.13
The characteristic lesions of established disease are tense and often intact blisters arising on normal or erythematous skin (Figs 4.41 and 4.42).
Generalized Vesicular Polymorphic Vegetans Nodularis Erythrodermic Seborrheic
Widespread
Cutaneous
Pretibial Brunsting-Perry
A
Localized
Mucous Membrane
Widespread
Mucosal
Desquamative Gingivitis
Localized
Oral
B
135 Bullous pemphigoid
They may measure up to several centimeters in diameter and are typically dome-shaped (Fig. 4.43). Often, they contain clear or bloodstained fluid. Any area of the body may be affected, but the blisters are most commonly located about the lower abdomen, the inner aspect of the thighs and on the flexural surfaces of the forearms, the axillae, and groin (Fig. 4.44).14 Grouping of lesions as seen in dermatitis herpetiformis is not usually a feature and symmetry is characteristically absent. A ‘cluster of jewels’ appearance of new blisters arising at the edge of resolving lesions as seen in linear IgA disease may, however, occasionally be a feature of bullous pemphigoid (Fig. 4.45).15 The lesions are often pruritic and a burning sensation is sometimes a feature. Nikolsky sign is usually negative. In contrast to mucous membrane pemphigoid, generalized bullous pemphigoid is not associated with scarring.
Reported mucosal involvement (frequently as ulcers) is highly variable, ranging from 8% to 58%.16–18 In a series of 115 patients, 24% had oral involvement and 7% had genital lesions.18 Lesions are found most often on the palate, the cheeks, lips, and tongue (Fig. 4.46). Other sites less commonly involved include mucosae of the nose, pharynx, conjunctiva and, rarely, the urethra and vulva (see below) (Fig. 4.47).17 In contrast to mucous membrane pemphigoid, mucosal involvement in generalized bullous pemphigoid is not associated with scarring.
Although bullous pemphigoid has been reported in association with a variety of internal malignancies, this may just be coincidental, merely reflecting the age incidence of these two diseases.19 In a series of almost 500 patients from Sweden, no increased incidence of cancer was observed.20 Other studies, however, have shown that there may be a positive correlation between internal malignancy and seronegative bullous pemphigoid patients.21 An association with neurological and psychiatric disorders, particularly multiple sclerosis and schizophrenia, has been demonstrated.22
Generalized bullous pemphigoid is a serious condition with a significant mortality ranging from 10% to 20%.1 Since the advent of steroid therapy and immunosuppressive agents, patients are more at risk of developing
136 Inherited and autoimmune subepidermal blistering diseases
severe iatrogenic disorders than of dying from their disease.23 Morbidity from this disease may be related more to the age and general state of health of the patient than to the severity of blistering.24 Although mortality from the disease is low, there has been a reported increase in mortality in the last 20 years of the twentieth century.25
It has been suggested that polymorphic pemphigoid is not an entity sui generis, but represents a potpourri of conditions including vesicular pemphigoid, linear IgA disease, and mixed subepidermal bullous disease in which patients show both linear IgG and linear IgA or dermal papillary granular IgA on direct immunofluorescence.31
Clinical variants of generalized pemphigoid Urticarial bullous pemphigoid presents with large persistent erythematous plaques, which sometimes display an annular or gyrate peripheral component (Figs 4.48 and 4.49).1 Rarely, small vesicles are also to be found.
Vesicular pemphigoid is a rare clinical variant in which the cutaneous manifestations show a striking overlap with dermatitis herpetiformis.26–29 Patients present with numerous small tense vesicles that may be symmetrical, intensely pruritic, and therefore associated with conspicuous excoriation.
Polymorphic pemphigoid is a somewhat confusing entity, which is similar to vesicular pemphigoid, but probably shows overlap with linear IgA disease.30–32 Patients present with burning and itching lesions predominantly affecting the extensor aspects of the limbs, back, and buttocks. Symmetry, grouping, and a polymorphic clinical appearance of papules, vesicles, and variably sized bullae, emphasize a similarity to dermatitis herpetiformis.
Pemphigoid vegetans is an exceedingly rare vegetative intertriginous variant that may be associated with chronic inflammatory bowel disease.33–40 Fewer than 10 cases have been documented. Patients present with vegetative, crusted, purulent, and sometimes eroded lesions in the groin, axillae, neck, hands, eyelids, inframammary, and perioral regions (Fig. 4.50). Vesicles and bullae may also be evident. Scarring has been described.40 The etiology of the vegetative lesions is unknown.
Seborrheic pemphigoid is a variant in which the clinical features are suggestive of pemphigus erythematosus.32
Pemphigoid nodularis represents the extremely rare association of lesions of bullous pemphigoid with intensely pruritic papules and nodules of nodular prurigo predominantly affecting the trunk and extremities (Fig. 4.51).41–43 The association of pemphigoid nodularis with immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome is the subject of a single case report.44
137 Bullous pemphigoid
A
B
Exceptionally, patients may show immunofluorescent evidence of bullous pemphigoid in the absence of clinical blistering.43 The cause of this unusual phenomenon is unknown, although in some patients at least, chronic scratching probably damages the basement membrane region with exposure of bullous pemphigoid antigens. There is a female predilection (2 : 1).43 The age range of this variant extends from 24 to 80 years but, as with classical bullous pemphigoid, the majority of patients are elderly.
Dyshidrosiform pemphigoid is a rare variant of pemphigoid in which patients develop 1–2-mm, tense ‘sago-grain-like’ vesicles on the palms and soles resembling dyshidrosiform dermatitis (pompholyx).45–51 Lesions may be localized, or precede or occur simultaneously with generalized disease. Overlap with pemphigoid nodularis has been described.52
Childhood pemphigoid exhibits lesions that are similar to their adult counterparts, but there is some tendency for lesions to be localized around the face, lower trunk, thighs, and genitalia, reminiscent of linear IgA disease in childhood (Fig. 4.52).7,8,53–62 Similarly, a ‘cluster of jewels’ appearance is sometimes evident.7 Palmar, plantar, and oral lesions are often present
138 Inherited and autoimmune subepidermal blistering diseases
• Brunsting-Perry pemphigoid – not a true variant of bullous pemphigoid – predominantly affects the head and neck and is associated with scarring.68
• Localized cutaneous nonscarring bullous pemphigoid (Eberhartinger, and Niebauer variant)69 predominantly affects the lower legs (in particular the pretibial region) of females. The former variant is considered in the section on mucous membrane pemphigoid. Although the latter nonscarring cutaneous form particularly affects the lower legs (Fig. 4.55), it may also present at a variety of other sites including forearms and hands, breasts, chest, buttocks, and umbilicus. Lesions in localized bullous pemphigoid may be related to trauma.69 It shows a peak incidence in the sixth decade. As with generalized bullous pemphigoid, patients present with tense, sometimes hemorrhagic, bullae that arise on normal or erythematous-appearing skin. Localized cutaneous nonscarring bullous pemphigoid is generally associated with a good prognosis.69
and may be the sole site of involvement in infants (Fig. 4.53). The mucous membranes may be affected, but scarring is absent. A number of children with primary localized penile and vulval lesions have also been described (Fig. 4.54).48,49,60,63,64 This is of particular clinical importance since it may be mistaken for evidence of sexual abuse. Childhood pemphigoid has a good prognosis and, as in adults, is usually self-limiting. Although the etiology is generally unknown, in some infant cases there appears to be a relationship to prior vaccination or immunization.60,65 Presentation during infancy is often severe, with blisters on the hands and feet.66 In this age group, the levels of anti-BP180 NC16A autoantibodies detected by ELISA are significantly higher than in adults with BP.66 Differences between childhood and infant cases have been described, but the importance of further subdividing this group is unclear.65
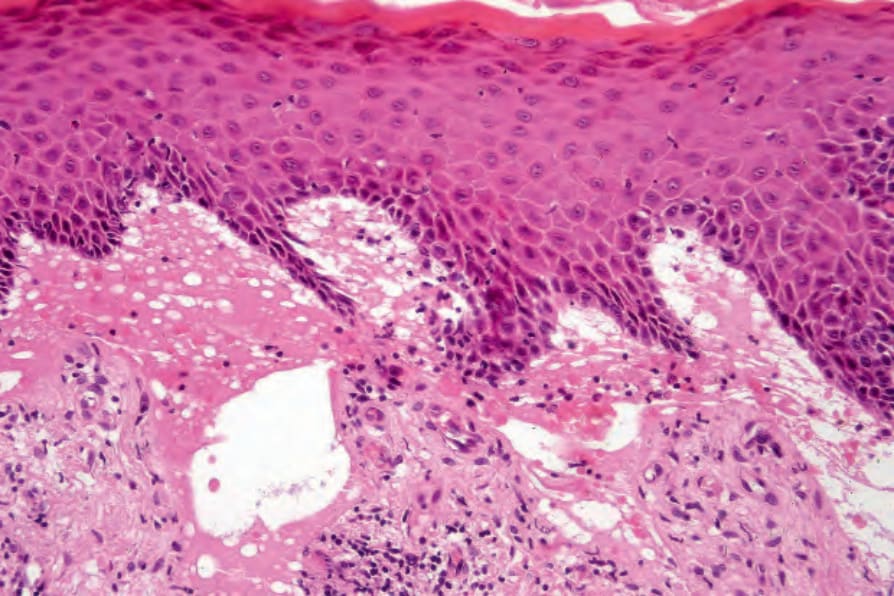
Fig. 4.38 Generalized severe recessive dystrophic EB: in addition to obvious subepidermal blistering there is dermal scarring and chronic inflammation.
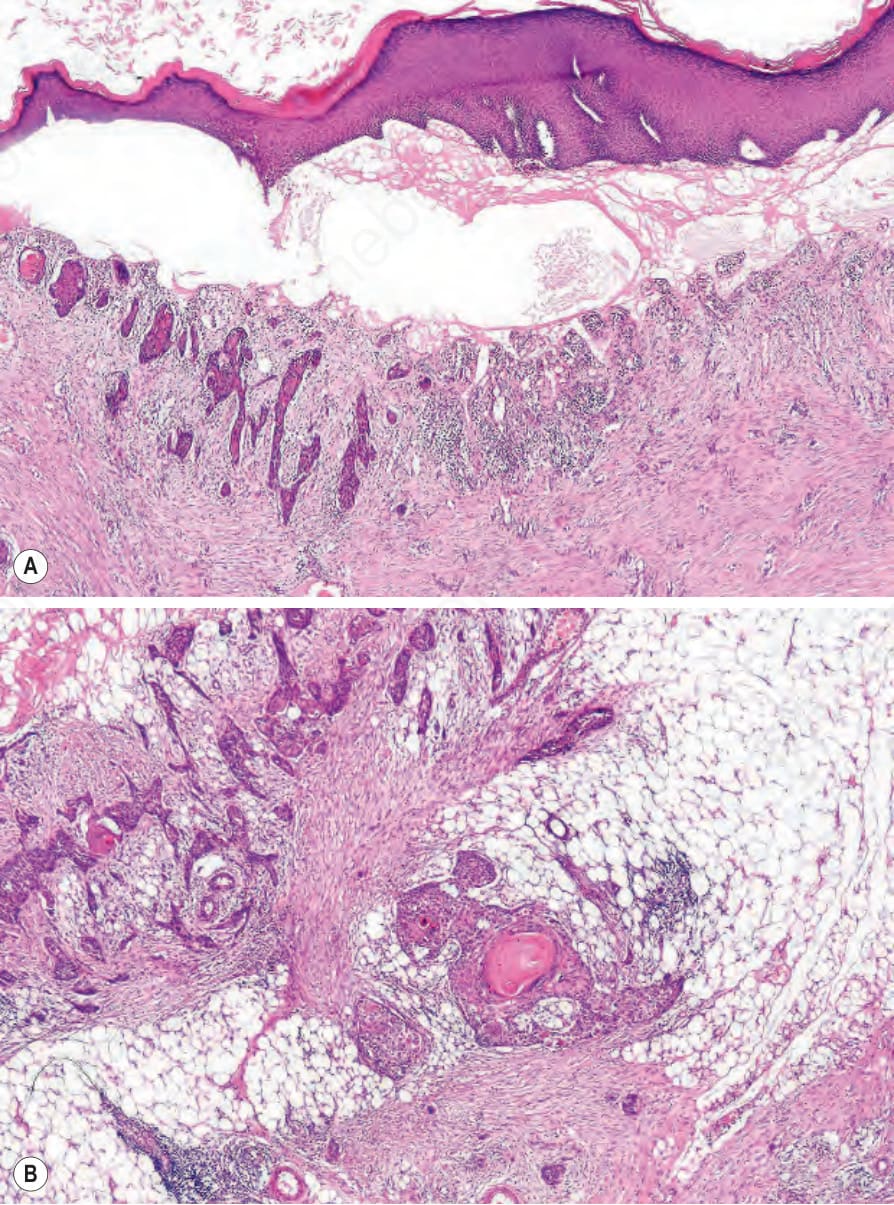
Fig. 4.39 (A, B) Generalized severe recessive dystrophic EB: biopsy from the forearm of a 30-year-old patient showing a cell-free subepidermal blister. In addition, a welldifferentiated squamous cell carcinoma extends into the subcutaneous fat.

Fig. 4.40 Bullous pemphigoid: classification.
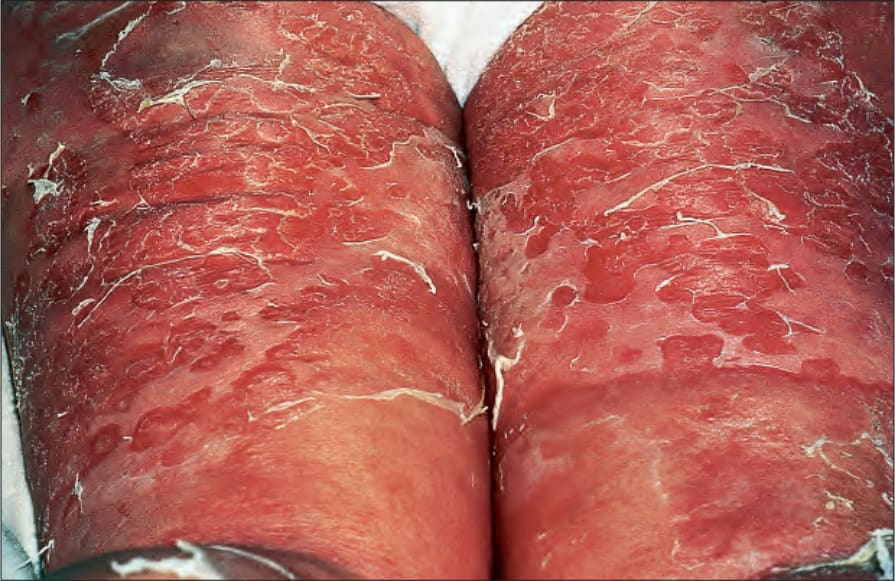
Fig. 4.41 Erythrodermic BP: blistering has developed against a background of generalized erythroderma. By courtesy of the Institute of Dermatology, London, UK.
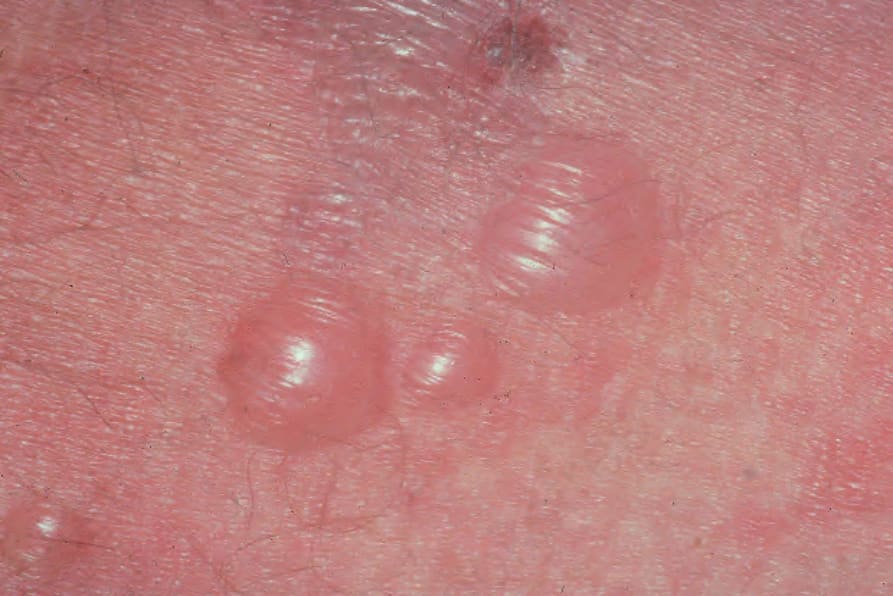
Fig. 4.42 BP: early tense blister arising on an erythematous base. By courtesy of the Institute of Dermatology, London, UK.
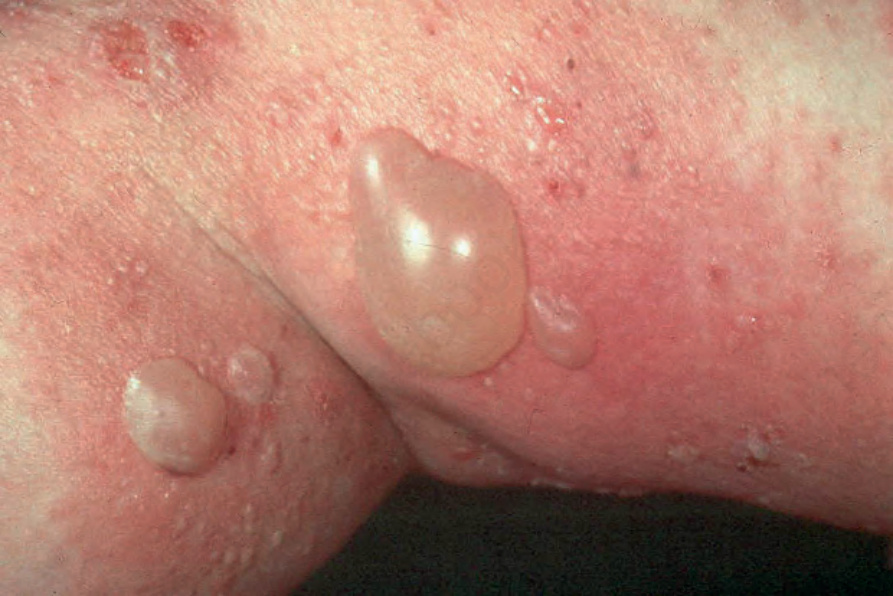
Fig. 4.43 BP: tense, dome-shaped blisters. The flexures are typically affected. By courtesy of the Institute of Dermatology, London, UK.
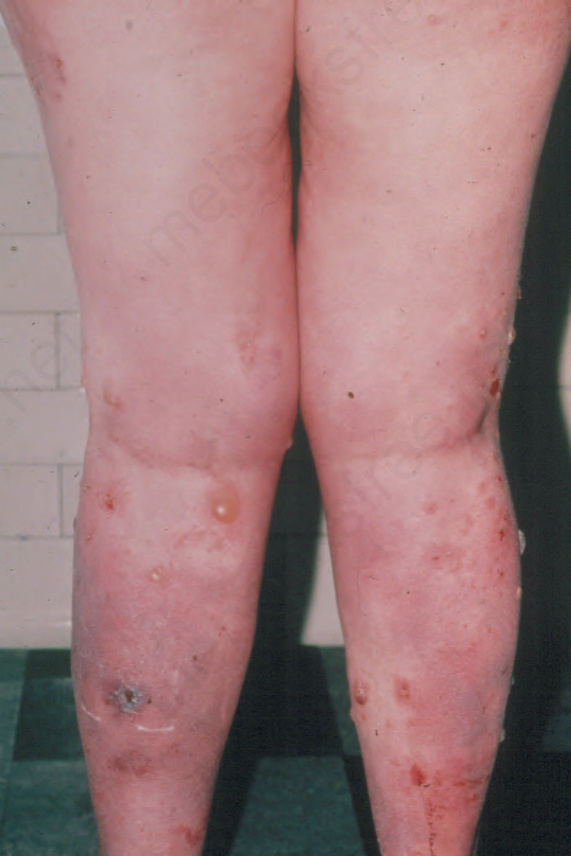
Fig. 4.44 BP: widespread, fluid-filled, hemorrhagic blisters on the arms of an elderly female. By courtesy of the late M. Beare, MD, Royal Victoria Hospital, Belfast, N. Ireland.
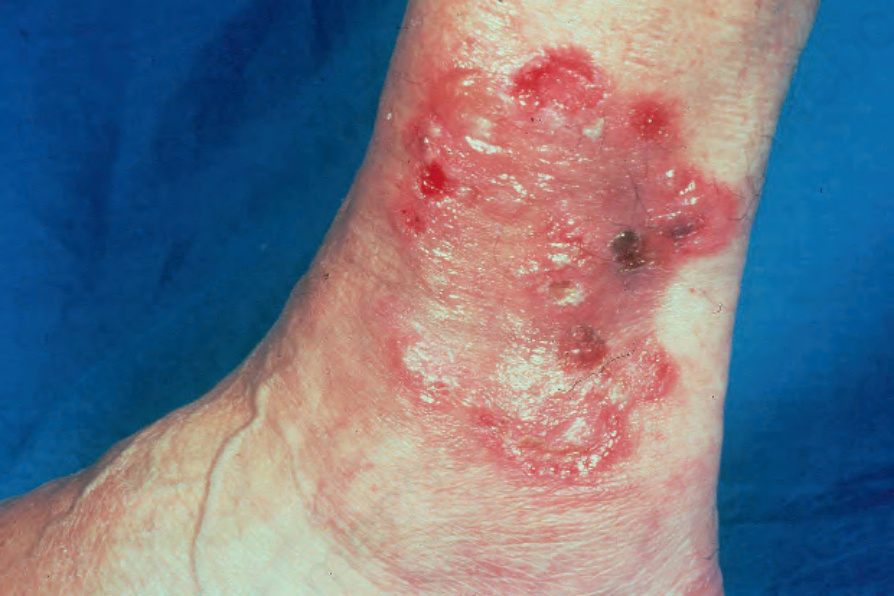
Fig. 4.45 BP: new blisters arising at the edge of a healing lesion (‘cluster of jewels’ sign). Although typically seen in childhood linear IgA disease, this is sometimes a feature of bullous pemphigoid. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
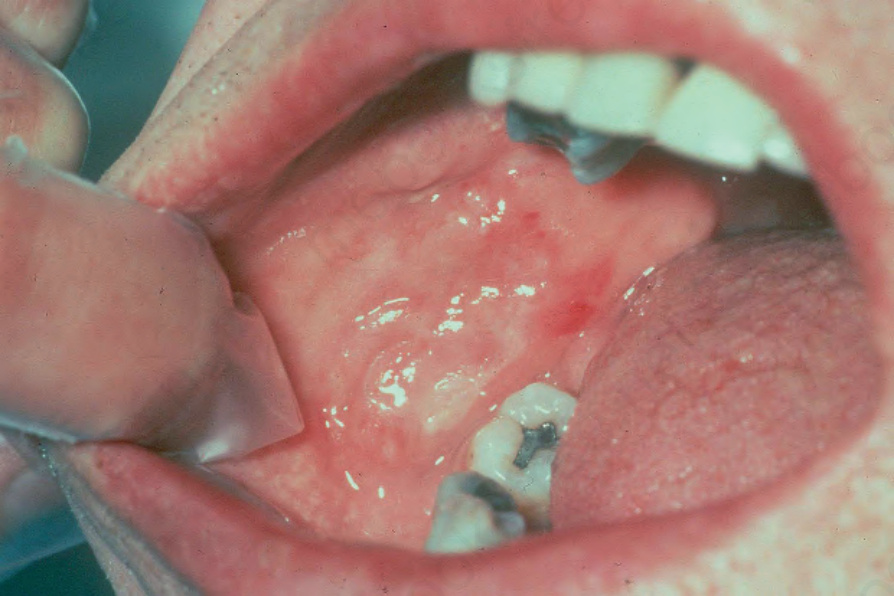
Fig. 4.46 BP: oral erosions are an occasional finding. Intact blisters are rare. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
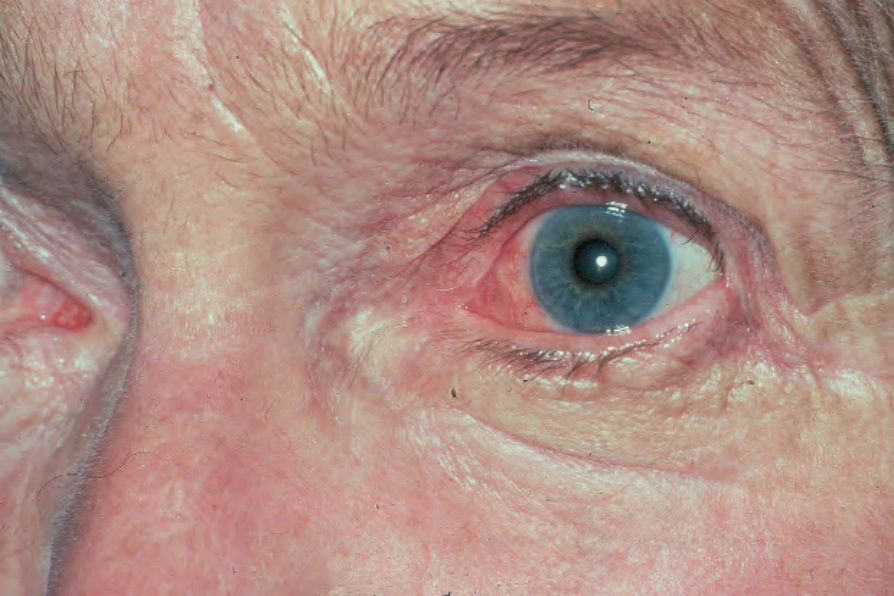
Fig. 4.47 BP: conjunctival injection is present. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
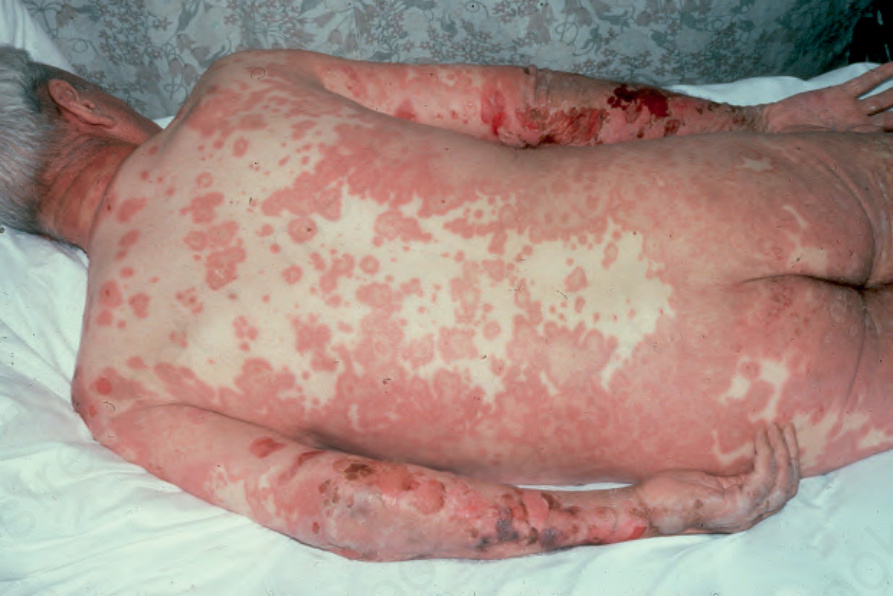
Fig. 4.48 Bullous pemphigoid: occasionally erythematous urticarial lesions may be the presenting feature. Blisters may not evolve until several weeks later. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
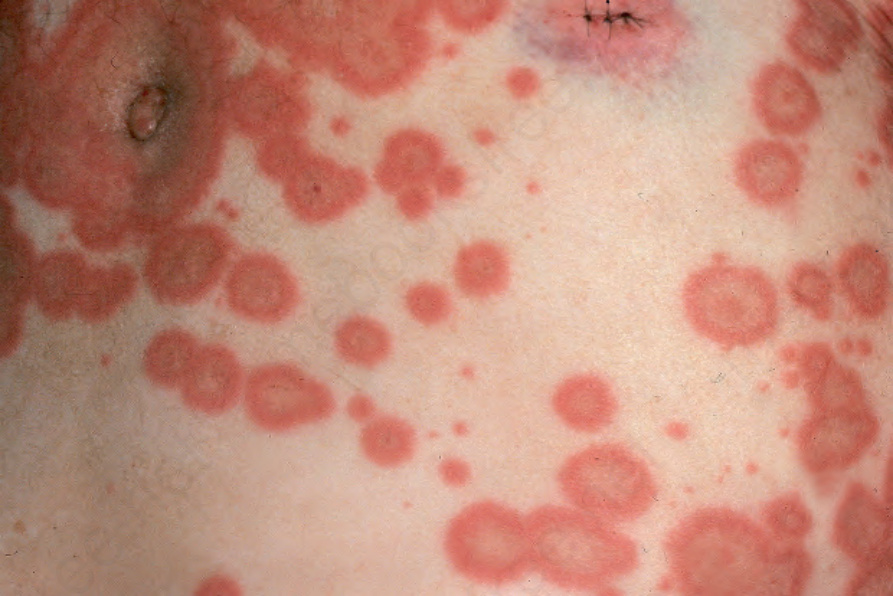
Fig. 4.49 Bullous pemphigoid: close up view. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
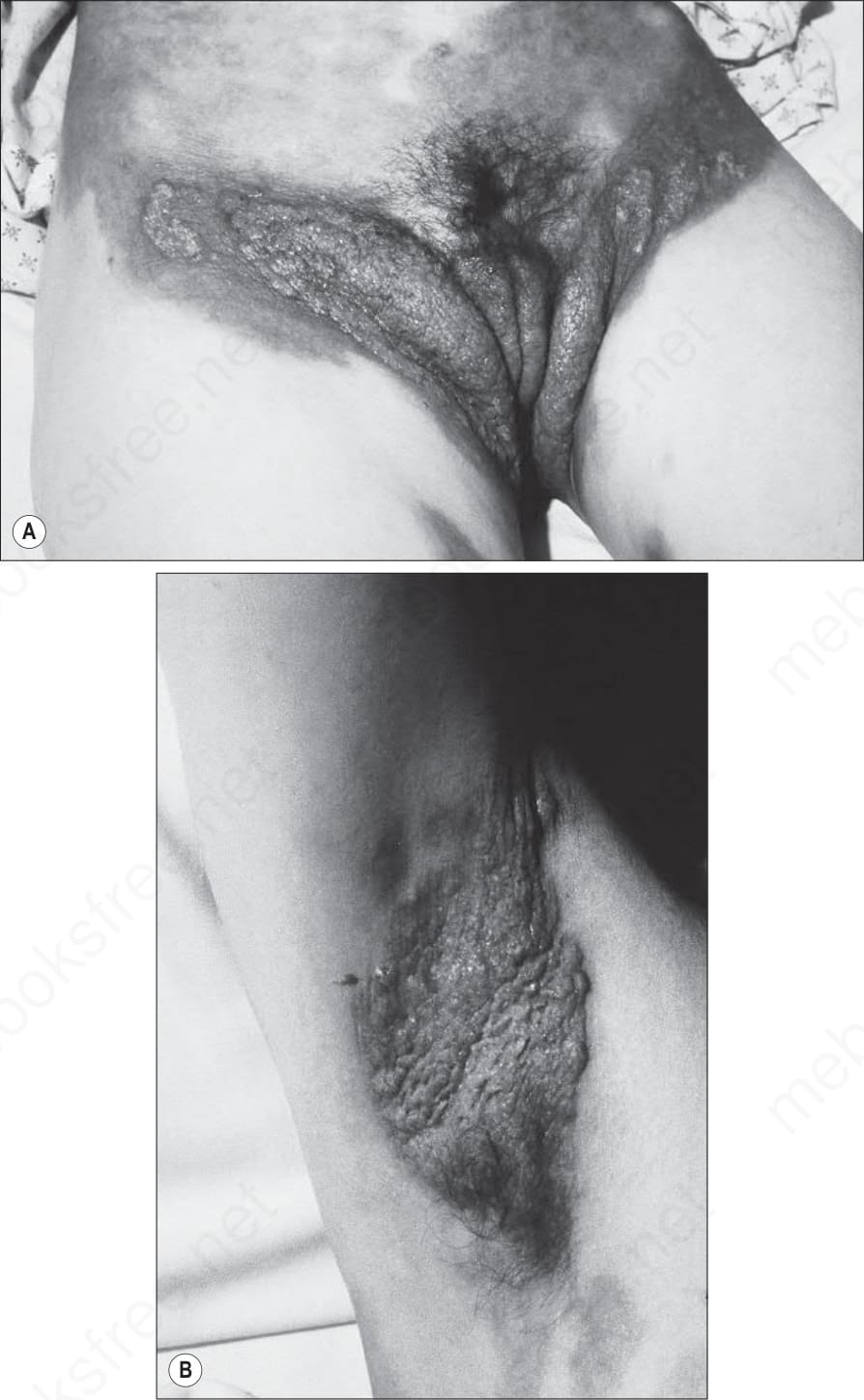
Fig. 4.50 (A, B) Pemphigoid vegetans: presentation as verrucous lesions in the flexures may result in considerable diagnostic difficulties. By courtesy of R.K. Winkelmann, MD, The Mayo Clinic, Scottsdale, Arizona, USA.
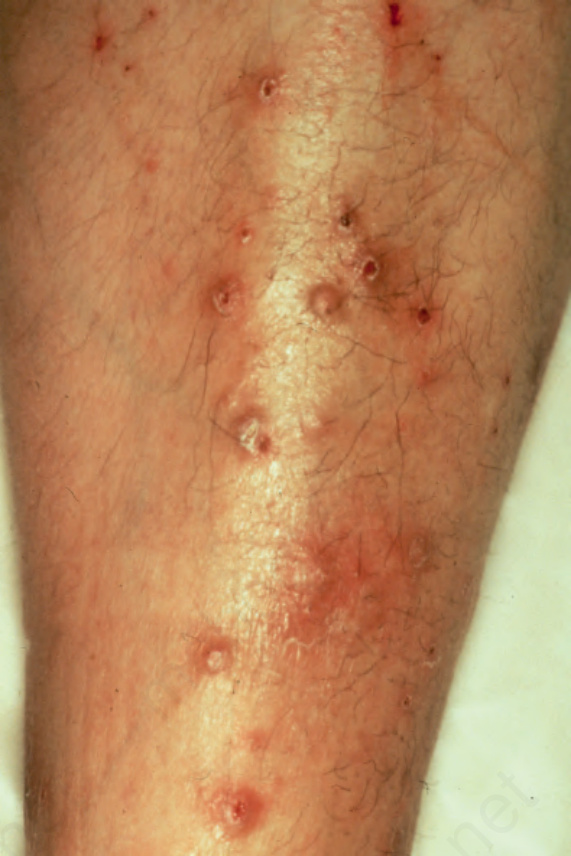
Fig. 4.51 Pemphigoid nodularis: in addition to bullous lesions, this patient also developed these pruritic nodules. By courtesy of H. Shimizu, MD, Keio University School of Medicine, Tokyo, Japan.
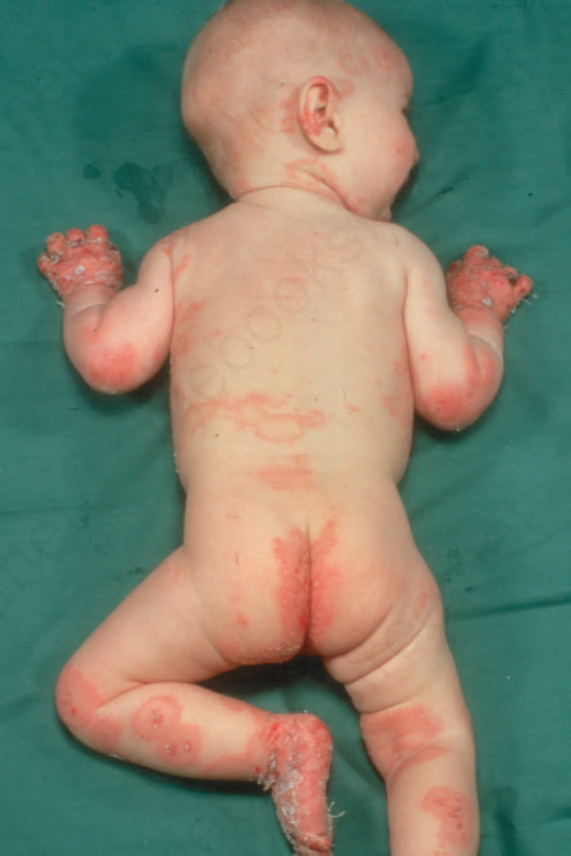
Fig. 4.52 Childhood BP: very rarely this disease affects young children and infants. There is a widespread distribution of bullae, which characteristically arise on an erythematous base. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
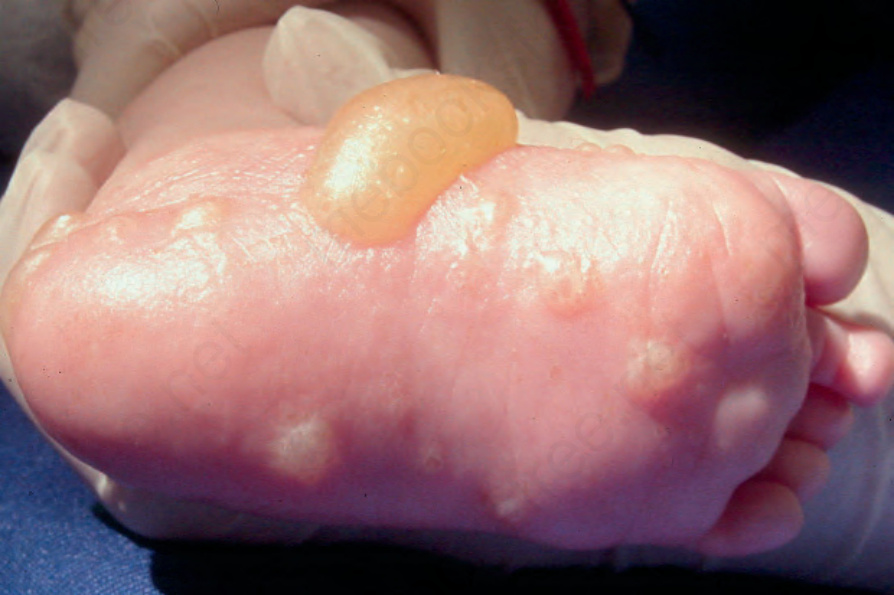
Fig. 4.53 Childhood BP: plantar involvement is sometimes the only site of disease. By courtesy of M. Liang, MD, The Children’s Hospital, Boston, USA.
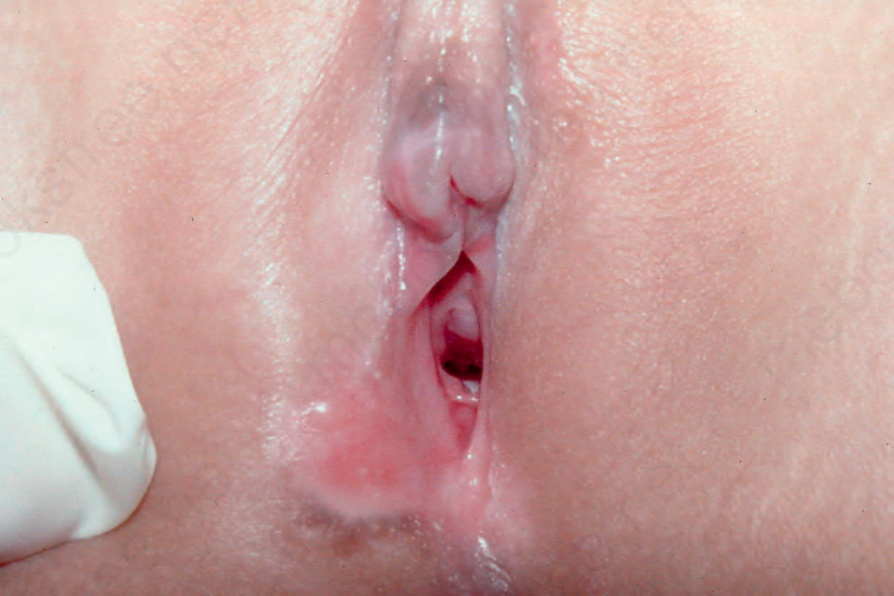
Fig. 4.54 Childhood BP: note the perineal scarring and isolated blister. By courtesy of M. Liang, MD, The Children’s Hospital, Boston, USA.
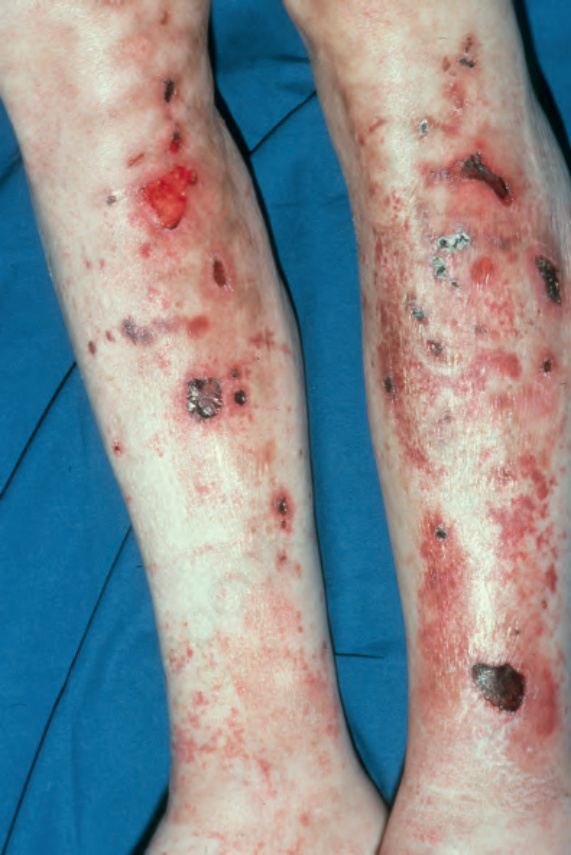
Fig. 4.55 Localized pemphigoid, nonscarring variant: lesions are found particularly on the lower legs of females. The prognosis is usually good, but occasionally the condition can become generalized. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
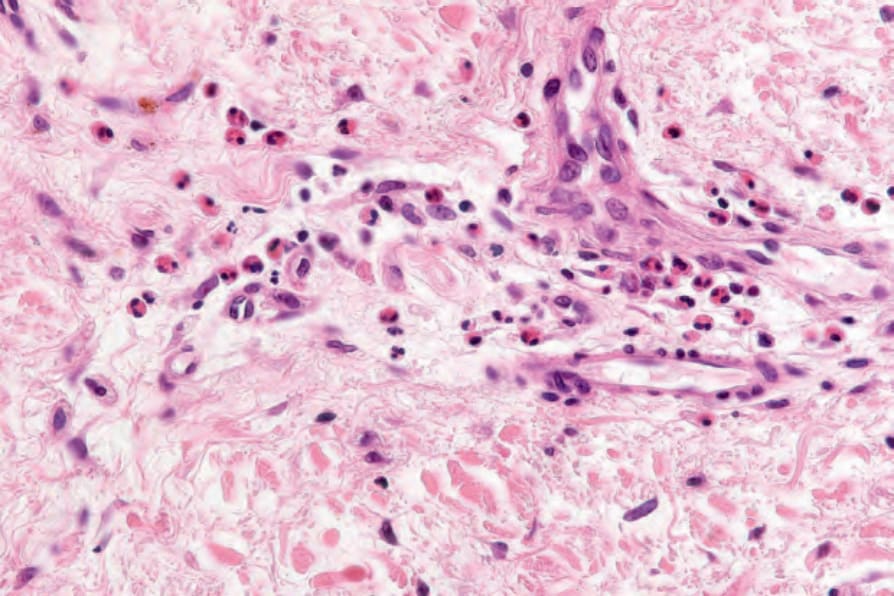
Fig. 4.58 Prebullous pemphigoid: there are numerous eosinophils.
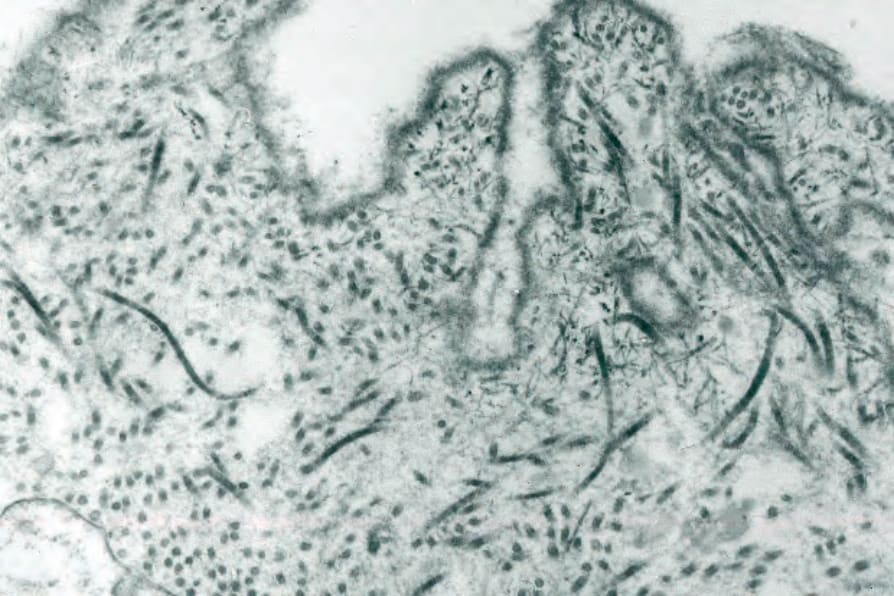
Fig. 4.70 BP: high-power view of the lamina densa.
Rare patients present with localized bullous pemphigoid at the site of trauma without much evidence of disease elsewhere.70
Mucosal pemphigoid/desquamative gingivitis Localized oral pemphigoid is described as a variant of desquamative gingivitis.71–73 The latter, of multifactorial etiology by definition, affects the marginal and attached gingivae. It shows a female predominance (9 : 1) and presents most frequently in the middle aged. Desquamative gingivitis may also be a manifestation of lichen planus and pemphigus.42 The diagnosis of localized oral pemphigoid depends upon the presence of a linear band of immunoreactants at the epithelial basement membrane region on direct immunofluorescence.71 Clinical features include erythema, edema, erosions, and ulcers.74 The oral lesions are nonscarring. Bullous pemphigoid-associated desquamative gingivitis may remain confined to the gingiva (the localized oral pemphigoid type), but approximately an equal proportion of patients develop full-blown cutaneous pemphigoid (Fig. 4.56).71
Localized cutaneous pemphigoid Although classical bullous pemphigoid not uncommonly presents initially as localized lesions that after a few months become generalized, occasional patients present with localized blisters that do not subsequently disseminate (localized bullous pemphigoid).67 Traditionally, this group has been subdivided into two variants:
Pathogenesis and histologic features The histologic features of bullous pemphigoid depend to some extent upon the age of the lesion biopsied. Early erythematous and urticarial lesions most
139 Bullous pemphigoid
often show upper dermal edema associated with a perivascular lymphohistiocytic infiltrate accompanied by usually conspicuous eosinophils (Figs 4.57 and 4.58). Eosinophilic spongiosis is sometimes evident and occasionally, if eosinophils are present in sufficient numbers, flame figures may be a feature. Mild interface changes characterized by basal cell hydropic degeneration can be seen in early or prodromal lesions.
If the biopsy is taken from an established blister, the changes are most often those of an inflammatory (cell-rich) variant.75 The blister, which is subepidermal, is typically unilocular and covered by attenuated epithelium (Fig. 4.59). In early lesions the roof epidermis may appear unaffected or show occasional to even confluent necrotic basal keratinocytes. The blister contents include coagulated serum, fibrin strands, and large numbers of inflammatory cells including conspicuous eosinophils (Fig. 4.60). Variable numbers of neutrophils may be present. In cases in which an old lesion is biopsied the blister may appear at least focally, at an intraepidermal level due to re-epithelization.
A typical finding in bullous pemphigoid is retention of the dermal papillary outline (festooning) which project like sentries into the vesicle cavity (Fig. 4.61). The underlying dermis is inflamed and usually shows widespread severe edema. An infiltrate of eosinophils and mononuclears surrounds the
140 Inherited and autoimmune subepidermal blistering diseases
blood vessels and extends between the adjacent collagen bundles. Leukocytoclasis is not seen and features of vasculitis are absent. The adjacent papillary dermis is often edematous and, very occasionally, eosinophil microabscesses are a feature (Fig. 4.62). Exceptionally rarely, neutrophil microabscesses may be seen (see vesicular pemphigoid), raising diagnostic confusion with dermatitis herpetiformis. Eosinophilic spongiosis is also sometimes evident in the adjacent epidermis (Fig. 4.63).76
Cell-poor (noninflammatory) features are occasionally seen if biopsies are taken from lesions arising on noninflamed skin (Fig. 4.64). Because inflammatory cells are sparse or, exceptionally, even absent in such cases, there may be considerable problems with the differential diagnosis, particularly if adequate clinical information and immunofluorescence findings are not available.
Vesicular/polymorphic pemphigoid is characterized by subepidermal vesicles with features suggesting either bullous pemphigoid or dermatitis herpetiformis or both (Fig. 4.65). Neutrophil dermal papillary microabscesses, which are often regarded as pathognomonic of dermatitis herpetiformis, may be seen in this variant (Fig. 4.66).
Pemphigoid vegetans is characterized by acanthosis, often with pseudoepitheliomatous hyperplasia, papillary dermal edema with subepidermal clefting or frank vesicle formation and an inflammatory cell infiltrate of eosinophils, lymphocytes, histiocytes, and occasional neutrophils.
Pemphigoid nodularis exhibits pruriginous lesions, which are characterized by hyperkeratosis and acanthosis, and which may amount to pseudoepitheliomatous hyperplasia and dermal fibrosis (Fig. 4.67). In the dermis, a perivascular infiltrate of lymphocytes and eosinophils is present. The blisters show typical features of bullous pemphigoid (Fig. 4.68).
Localized nonscarring (pretibial) bullous pemphigoid usually shows the histology of cell-rich bullous pemphigoid. Localized oral pemphigoid is typified by a subepithelial vesicle (when present) and cannot be distinguished histologically from oral involvement in mucous membrane pemphigoid (see below).
Ultrastructurally, in early lesions of bullous pemphigoid, the dermal– epidermal cleavage is seen to have developed between the plasma membrane of the basal keratinocyte and the lamina densa, through the lamina lucida.77
141 Bullous pemphigoid
A
B
A
B
142 Inherited and autoimmune subepidermal blistering diseases
BP180 autoantibodies correlate not only with the disease severity, but also with the formation of erythematous and urticarial lesions in BP.93 Serum levels of IgG1 and IgG4 targeting the noncollagenous 16A domain of BP180 parallel disease activity and predict a more aggressive clinical behavior.94
Split skin indirect studies are essential in the investigation of a patient in whom a linear IgG antibasement membrane antibody has been detected.95–97 Such antibodies are also characteristic of mucous membrane pemphigoid, herpes (pemphigoid) gestationis, inflammatory epidermolysis bullosa, and bullous systemic lupus erythematosus. The antibodies in pemphigoid variants (with the exception of the anti-p105 and anti-p200 variants discussed below) bind to the epidermal side of 1 M NaCl-split skin whereas those of inflammatory epidermolysis bullosa and bullous systemic lupus erythematosus bind to the floor.
The lamina densa is therefore located along the floor of the blister (Figs 4.69 and 4.70). Degenerative changes in the basal cells, including villous process formation, mitochondrial swelling, and cytoplasmic vacuolization, are frequently found. Hemidesmosomes may appear reduced in number or may even be absent.78 Intercellular edema between adjacent basal cells is a common finding.79 If specimens from established inflammatory lesions are examined, the lamina densa is sometimes fragmented or entirely absent.49
In those patients in whom indirect fluorescent studies are not available, similar information may sometimes be obtained through the localization of lamina densa constituents, such as type IV collagen or laminin-1, using paraffin-embedded direct immunoperoxidase techniques. In pemphigoid, the staining is found along the floor of the blister, whereas in inflammatory epidermolysis bullosa and bullous systemic lupus erythematosus it is located along the roof (see Figs 4.7 and 4.8). In addition, a recent study demonstrated an immunofluorescence pattern in adnexal structures similar to that observed in the epidermis.98 Besides serum, saliva has been reported to represent a convenient alternative for detection of BP180 NC16a autoantibodies.99
Bullous pemphigoid is characterized by a linear antibasement membrane zone antibody using the indirect immunofluorescent technique.80 Although IgG is invariably present (and most commonly of the IgG4 subclass), other immunoglobulins, including IgE, may be represented.81 Such antibodies are present in around 75–80% of patients.82–85 Sensitivity can, however, be increased to 90% if split skin is used as substrate.18 Although earlier studies failed to detect a relationship between the antibody titer and disease activity or severity, more recently it has been shown that serum antibodies to the NC16A domain of BP180 (a subunit of the bullous pemphigoid antigen) do correlate with disease activity (see below).86–90 In addition, autoantibodies against BP180, and to a lesser degree to BP230, correlate with the clinical course of topically treated BP patients and can be used to monitor the effectiveness of the treatment.91 Furthermore, while autoantibodies in patients with the inflammatory variant of BP generally target the NC16A domain of BP180, autoantibodies in patients with noninflammatory variant usually react with the midportion of collagen XVII.92 Serum titers of IgE anti
Bullous pemphigoid antibodies are capable of complement fixation in as many as 75% of patients.100,101 Most of complement fixation in bullous pemphigoid antibody resides in the IgG4 subclass.102
Linear in vivo-bound immunoglobulin at the dermal–epidermal interface on direct immunofluorescence is present in 90% or more of patients (Fig. 4.71).18,103 Complement (C3) is also usually present and is sometimes the sole immunoreactant (Fig. 4.72).104 Other immunoglobulin subclasses including IgM, IgA, and IgE may be detected occasionally.85,100,105 In addition to C3, the other components of the classical complement pathway, in particular C5b-9 (the membrane attack complex) and members of the alternative complement pathway, including properdin, factor B, and B-1H-globulin, may also be identified.85,106 There is, therefore, evidence that both the
143 Bullous pemphigoid
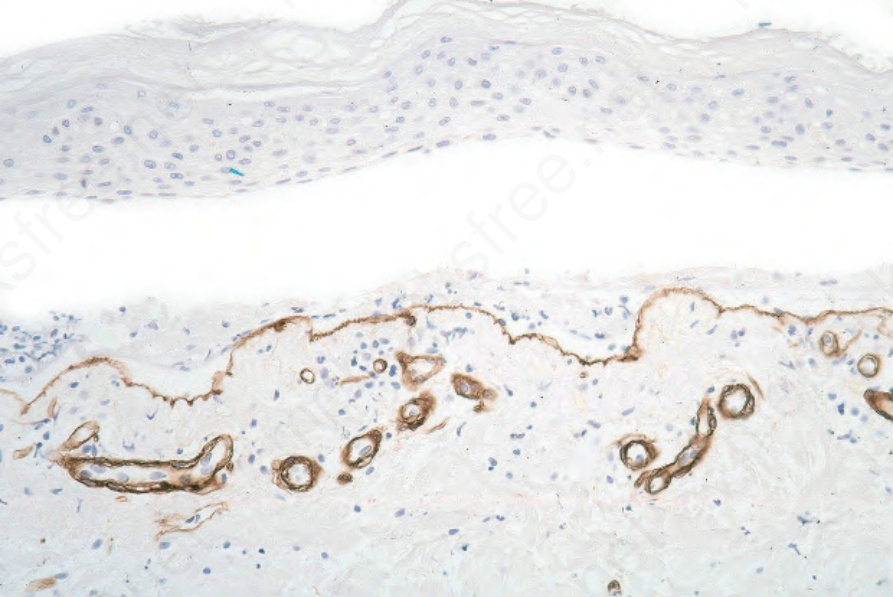
Fig. 4.7 Paraffin-embedded immunoperoxidase antigen mapping: in bullous pemphigoid, type IV collagen is present along the floor of the blister.
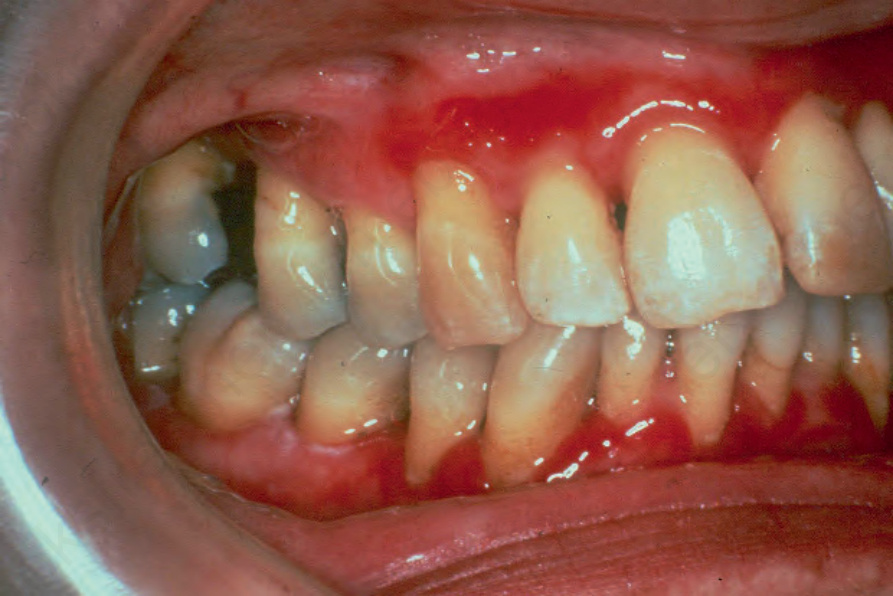
Fig. 4.56 Desquamative gingivitis: note the intense gingival erythema and retraction. Such features may also be seen in mucous membrane pemphigoid and pemphigus. By courtesy of P. Morgan, FRCPath, London, UK.
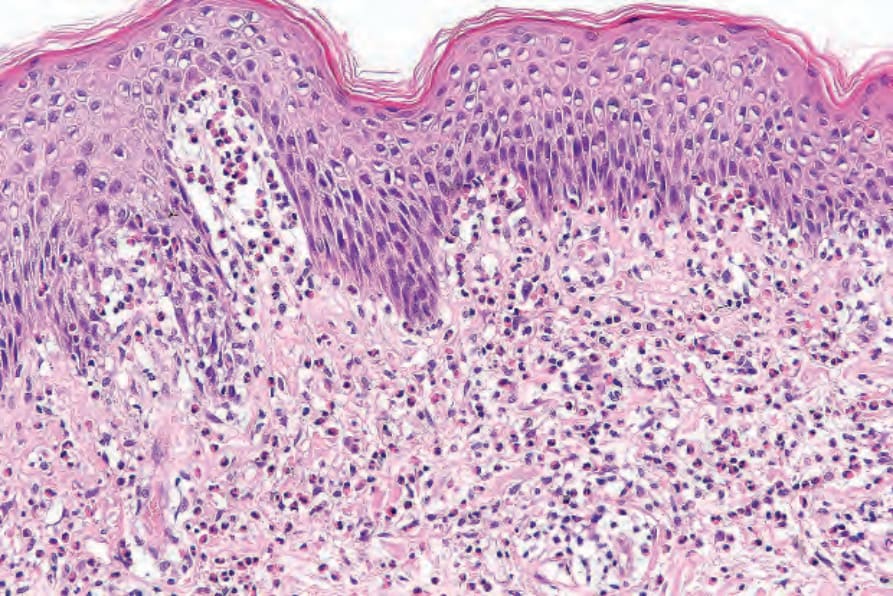
Fig. 4.57 Prebullous pemphigoid: there is upper dermal edema and a perivascular lymphohistiocytic infiltrate with conspicuous eosinophils.
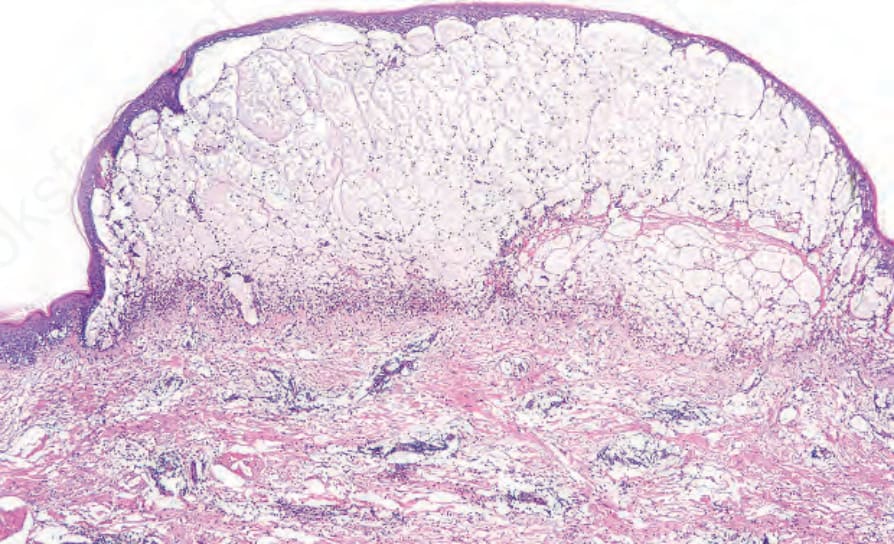
Fig. 4.59 BP: an established lesion showing a subepidermal tense, dome-shaped blister containing edema fluid, fibrin, and inflammatory cells.
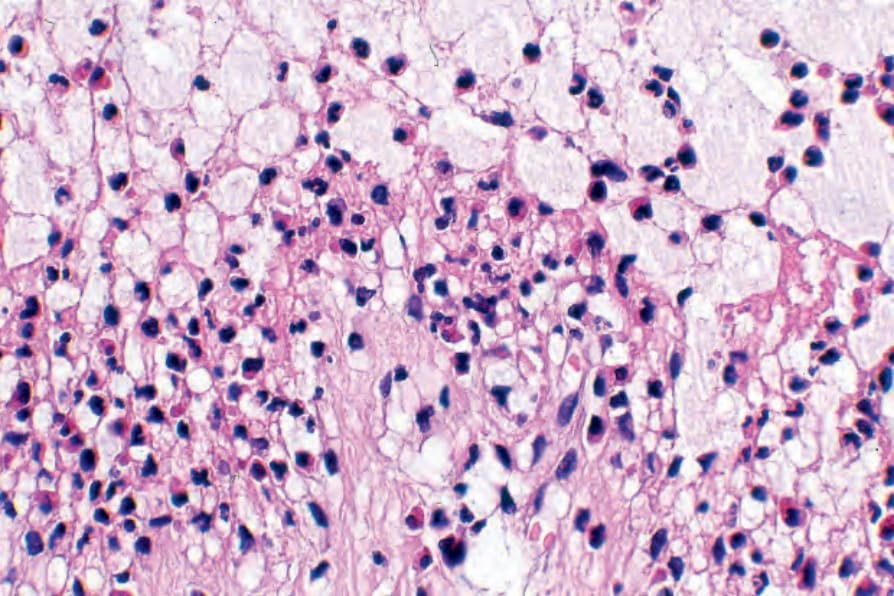
Fig. 4.60 BP: the blister cavity contains large numbers of eosinophils.
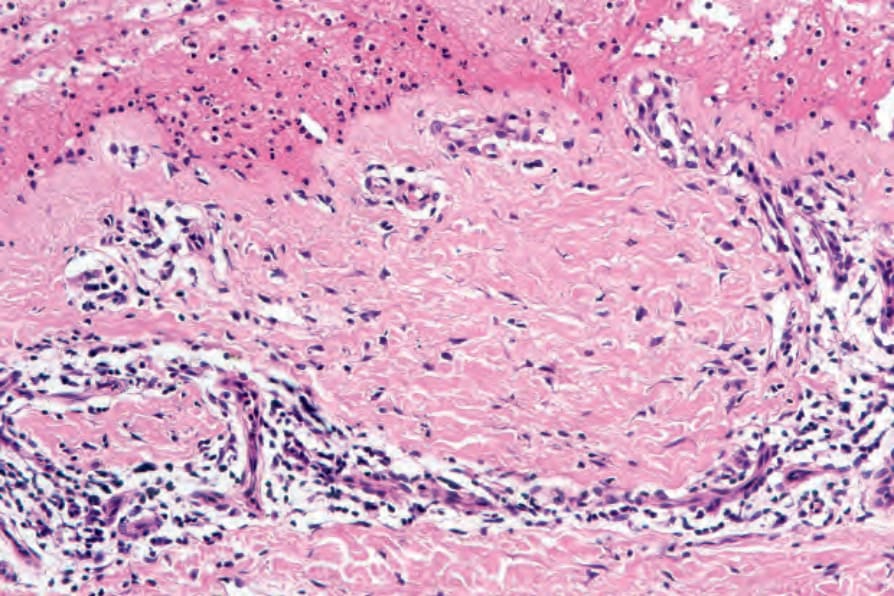
Fig. 4.61 BP: preservation of the dermal papillary outline (festooning) is a characteristic feature.
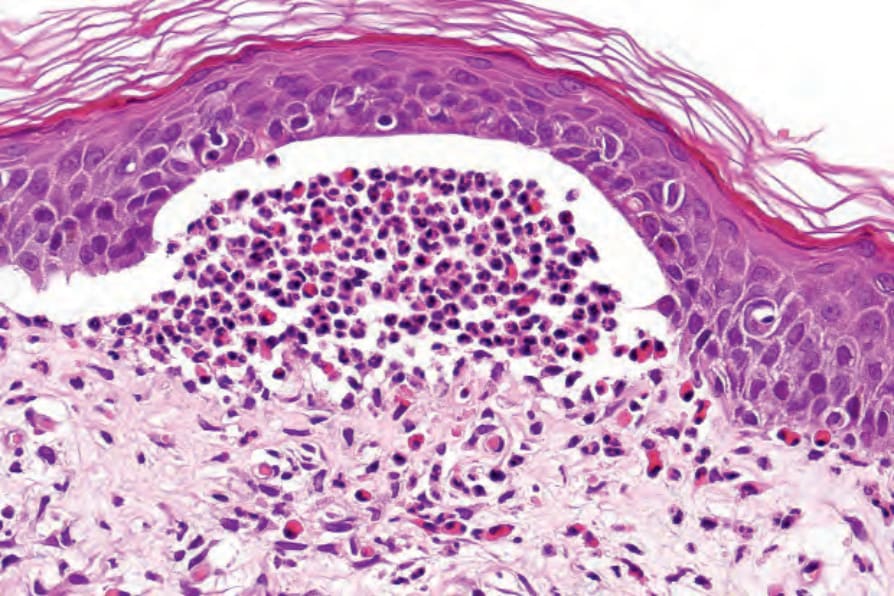
Fig. 4.62 BP: the presence of eosinophil microabscesses in the dermal papillae is a useful although rare diagnostic marker.
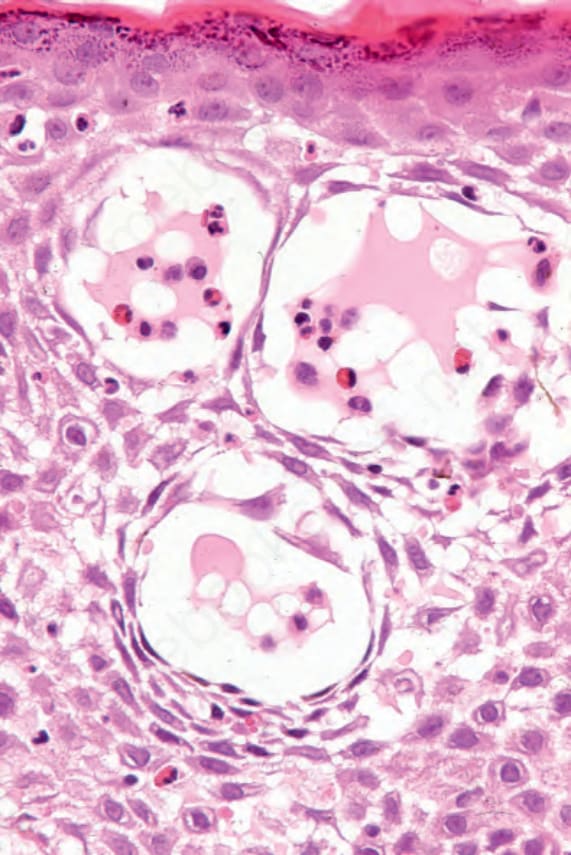
Fig. 4.63 BP: eosinophilic spongiosis is sometimes seen in the epidermis adjacent to the blister.
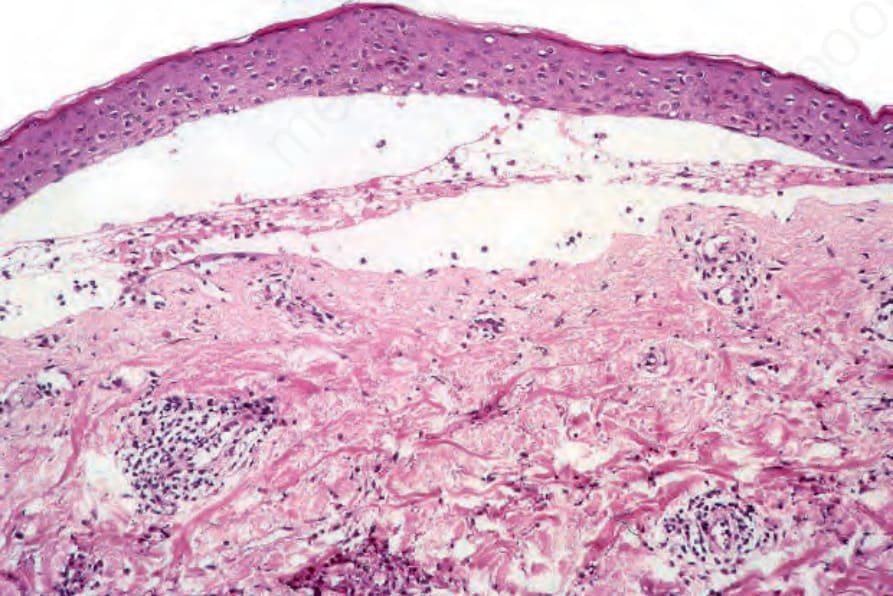
Fig. 4.64 Cell-poor pemphigoid: this is a very uncommon variant and is most often seen if a very early lesion is sampled. The blister contains only a little edema fluid and there is a light chronic inflammatory cell infiltrate in the superficial dermis.
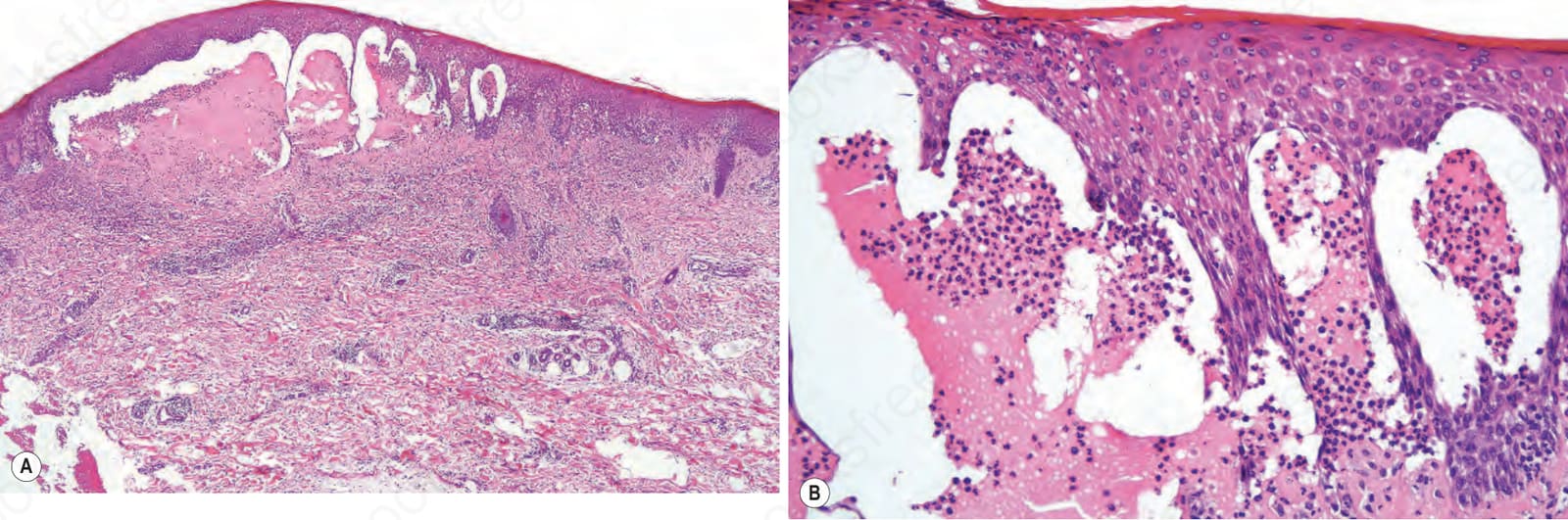
Fig. 4.65 Vesicular pemphigoid: (A) low-power view showing a multilocular blister; (B) the blister contains a neutrophil-rich infiltrate.
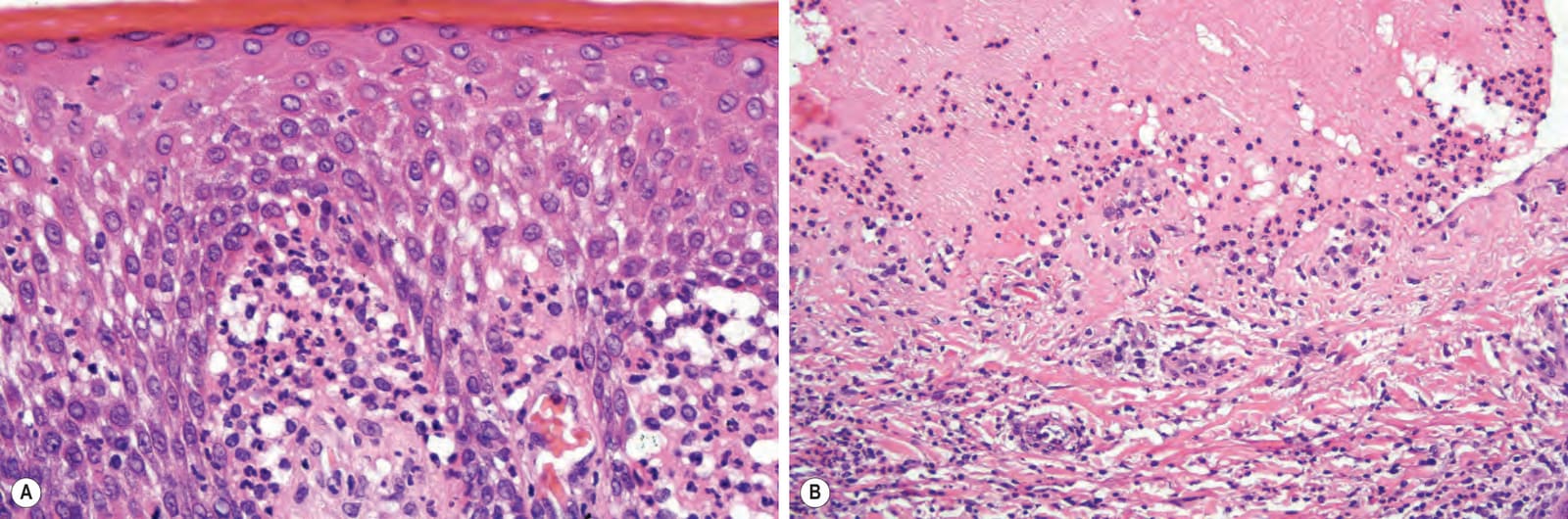
Fig. 4.66 Vesicular pemphigoid: (A) neutrophil microabscesses in the adjacent dermal papillae heighten the resemblance to dermatitis herpetiformis. It would be impossible to establish the diagnosis of bullous pemphigoid without appropriate immuno-fluorescent findings; (B) preservation of the dermal papillae may be a clue to the correct diagnosis of pemphigoid.
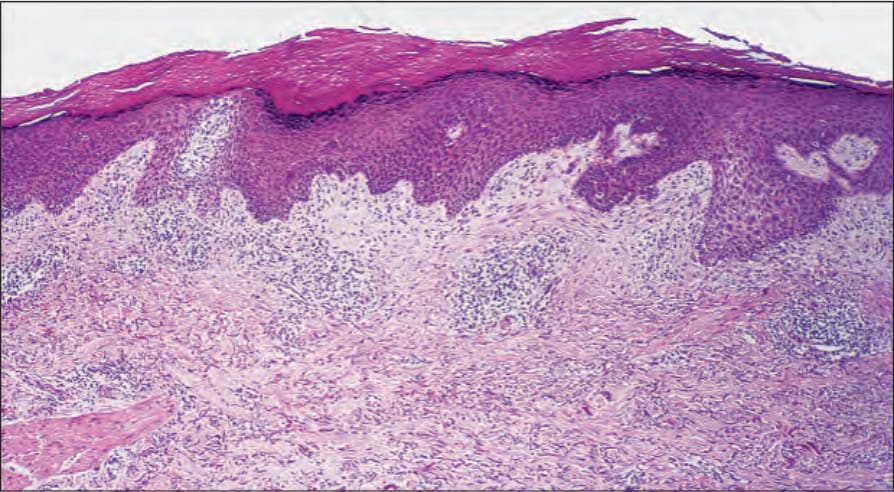
Fig. 4.67 Pemphigoid nodularis: this is a biopsy of a pruritic nodule showing hyperkeratosis, irregular acanthosis, dermal chronic inflammation, and scarring.
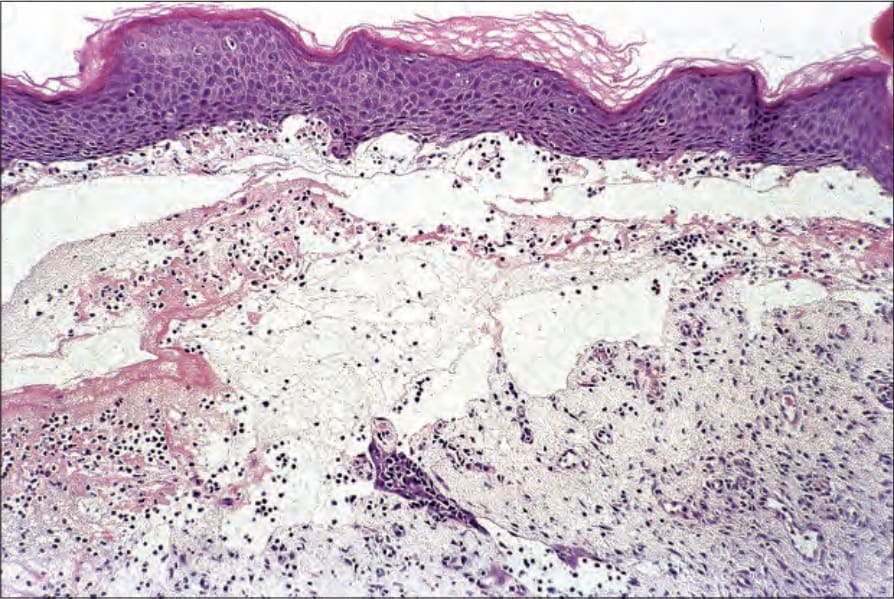
Fig. 4.68 Pemphigoid nodularis: this subepidermal blister comes from the same patient as shown in Fig. 4.67. Pemphigoid nodularis is of particular importance because the nodular lesions may precede clinical evidence of blistering.
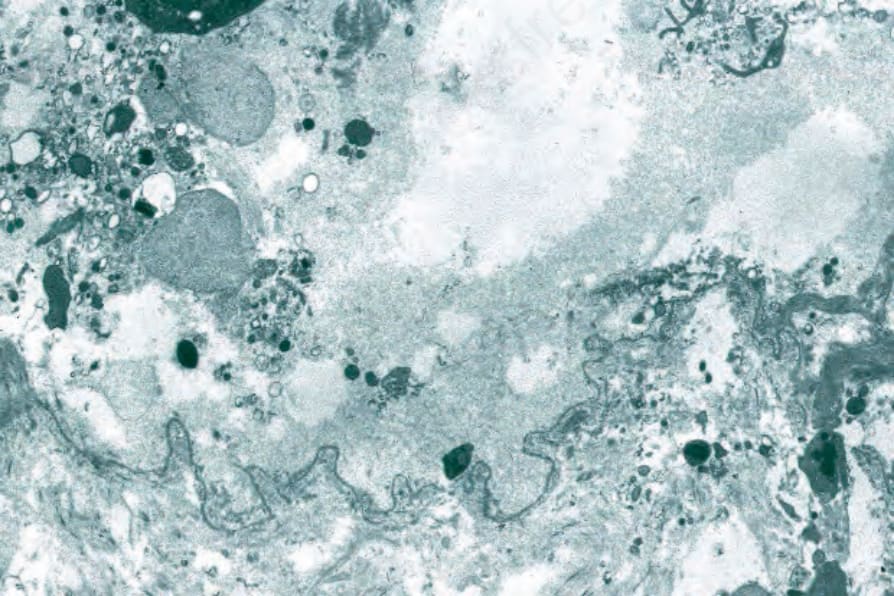
Fig. 4.69 BP: electron micrograph showing the lamina densa lying along the floor of the blister cavity.
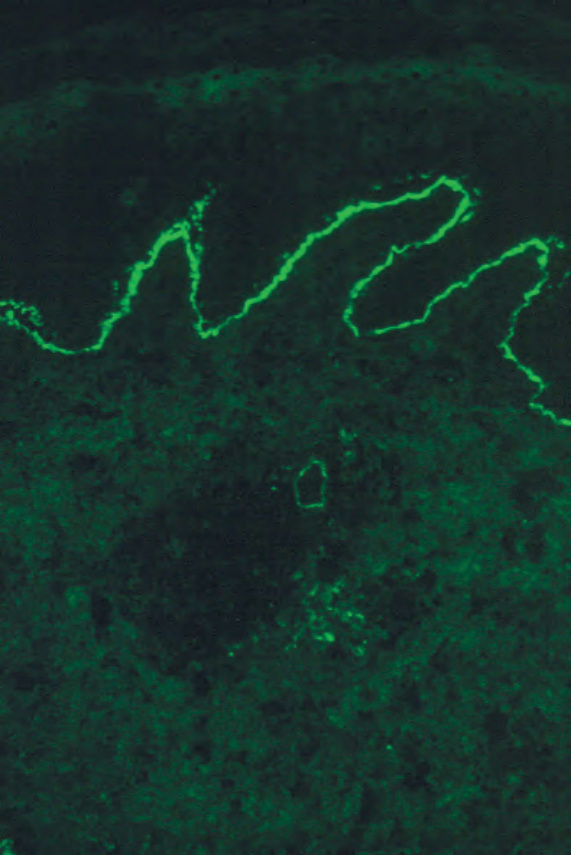
Fig. 4.71 BP: direct immunofluorescence of perilesional skin showing intense linear basement membrane zone staining (IgG).
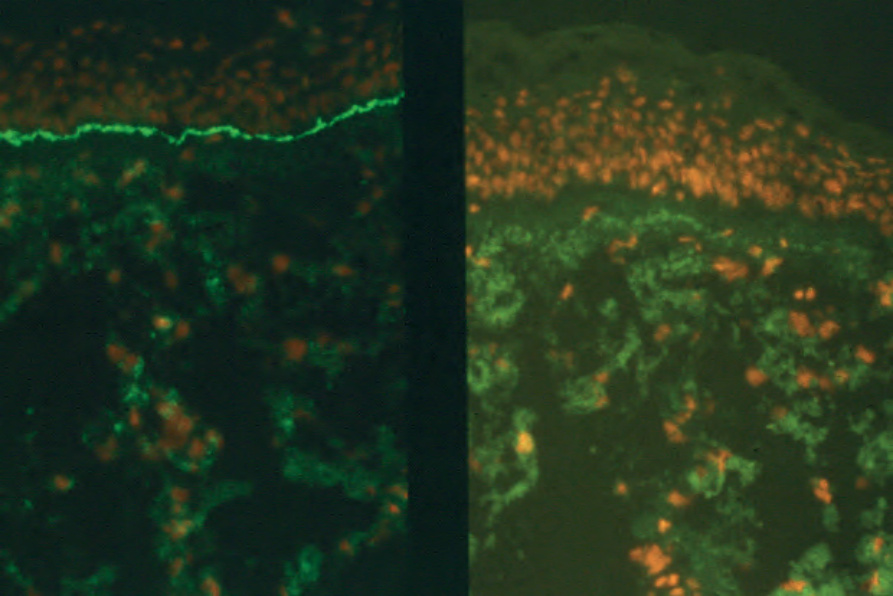
Fig. 4.72 BP: direct immunofluorescence showing C3 deposition (left), no staining is seen in the negative control (right). By courtesy of B. Boghal, FIMLS, Institute of Dermatology, London, UK.
classical and alternate complement pathways are involved in the pathogenesis of bullous pemphigoid.107 The classical complement pathway, however, predominates. A recent mouse model underscores the necessity of an intact innate immune system, as the depletion of complement or neutrophils or blockage of mast cell activation prevents blister formation.108
The immunofluorescence findings in erythematous, pruritic, urticarial, and eczematous prodromal lesions and childhood, dyshidrosiform, vesicular, nodular, and vegetans variants are similar to those seen in the conventional generalized disease.26–29,33–50,109,110 In polymorphic pemphigoid, either linear IgG or IgA deposits may be identified along the basement membrane region.30–32 The serum may contain either IgG or IgA antibodies.31
Immunofluorescence findings in localized cutaneous disease are variable. In some reports, patients show positive direct immunofluorescence for IgG and C3 at the dermal–epidermal junction and a positive indirect immunofluorescent test for bullous pemphigoid antibody, while others may be positive for in vivo-bound complement, but negative on indirect examination.68,69,110 One series has shown that almost 70% of patients with localized pemphigoid have circulating IgG antibodies in their sera and the presence of these can be relevant for serum-based testing, as discussed below.69,111 A caveat is that in one study, antibodies were also detected in more than half of normal subjects who did not subsequently develop the disease.112,113 This finding is further discussed below.
By direct immunoelectron microscopy, the immunoreactants (IgG and C3) are seen to be located within the hemidesmosomal plaque and upper lamina lucida (Fig. 4.73).114–118 Indirect immunoelectron microscopic studies show that the bullous pemphigoid antigen is most often detected intracellularly in the region of the cytoplasmic face of the hemidesmosome (Fig. 4.74).115,119–121
BP180 (collagen type XVII) is the major pathogenic antigen in bullous pemphigoid. The BPAG2 (COLI7A1) maps to the long arm of chromosome 10, locus 10q24.3.132 It is a transmembrane adhesion molecule comprising an intracytoplasmic N-terminal fragment, a transmembrane region, and a collagenous extracellular C-terminal ectodomain.138 The latter constitutes part of the anchoring filament and distally merges with the lamina densa. The antibodies directed against BP180 in bullous pemphigoid most commonly react with a short extracellular noncollagenous locus – NC16A (regions MCW0-MCW3) – located within the upper lamina lucida proximal to the collagenous segment (Fig. 4.76).138–141 It now appears that antibodies specific to this area are generally required for blister formation and, while antibodies may also target BP180 non-NC16A domains, these latter antibodies do not appear to be pathogenic in most cases.135–137 This finding reconciles the fact that antibodies to both BP180 and BP230 can be seen in a significant portion of the population without blister formation as these are not against the critical NC16A region of BP180.86
The immunoelectron microscopic observations in childhood bullous pemphigoid, vesicular pemphigoid, polymorphic pemphigoid, pemphigoid nodularis, pemphigoid vegetans, and localized pemphigoid are identical to those of classic bullous pemphigoid.122,123
Two principal bullous pemphigoid antigens are recognized by Western blot and immunoprecipitation studies: one is 230 kD (BPAG1) and the other is approximately 180 kD (BPAG2) (Fig. 4.75).124–130 These represent products of distinct genes.131–134
BP230 maps to the short arm of chromosome 6, locus 6p11-12.132 It belongs to the plakin family and shows homology with plectin and the desmogleins.133 It is wholly intracellular and localizes to the hemidesmosome. BP230 is not involved in the early stages of the pathogenesis of blistering but is of importance as a secondary event; antibodies against this antigen are not required for blister formation in most cases.135–137
Between 50% and 90% of patients with generalized bullous pemphigoid have antibodies that react with BP230 and 35–50% have antibodies that
144 Inherited and autoimmune subepidermal blistering diseases
Circulating antibodies against BP180 or BP230 have also been defined in many of the other less common variants of bullous pemphigoid, including localized and vesicular forms, pemphigoid vegetans, erythrodermic pemphigoid, and pemphigoid nodularis.142,146–150
In childhood pemphigoid, the antibodies also react against the same antigens.151 In addition, rarely there may also be antibodies that react with the linear IgA 120-kD antigen.151 The BP180 antigen is most often targeted, and immunoblot analyses have shown that the antibodies react specifically with the NC16A domain as in adult patients. In some children at least, the IgG subclasses differ from adult disease, consisting of all IgG subclasses or IgG2 in isolation.18 IgE antibodies are not a feature of childhood disease.
More recently, two patients with a nonscarring, bullous pemphigoid-like illness characterized by neutrophil-rich subepidermal blisters resembling dermatitis herpetiformis and antibodies to a unique 105-kD protein – so-called anti-p105 pemphigoid – have been documented.152–154 This antigen localizes to the dermal side of split skin on indirect immunofluorescence. Its precise nature has not yet been determined.
Anti-p200 pemphigoid is characterized by antibodies to a lower lamina lucida basement membrane antigen.155–157 Patients generally present with a nonscarring bullous pemphigoid-like illness although linear IgA disease-like and dermatitis herpetiformis-like variants have also been reported.155 The disease has also been described in association with psoriasis vulgaris and only rarely with pustular psoriasis.156,158,159 With split skin IIMF, the antibodies bind to the floor of the blister cavity.155 With indirect immunoelectron microscopy, the antibodies bind to the lower lamina lucida.160,161 It has been suggested, that antibodies against laminin γ1 have a pathogenetic role and that this molecule represents the autoantigen in the development of anti-p200 pemphigoid.162,163
Basal keratinocyte
HD plaque
NH2
Globular cytoplasmic domain
Anti-p450 pemphigoid has been documented in a single patient. The antigen, which has been localized to the basal keratinocyte, belongs to the plectin family.164 Its precise nature has yet to be determined.
Exceptionally, bullous pemphigoid may be associated with antiplectin antibody.165
Bullous pemphigoid has been described following PUVA therapy for mycosis fungoides. A case arising in the setting of radiation therapy and one in the setting of photodynamic therapy have also been noted, perhaps suggesting a role for tissue damage in the pathogenesis of this disease.166,167
Cell membrane NC16a
Transmembranous domain
Rod-like interrupted collagenous domain
Lamina lucida
COL1
Flexible ‘tail’
A mechanism for blister development in bullous pemphigoid has been proposed by Jordon et al.82,168 and is outlined as follows. Following antibody–antigen interaction and complement fixation, various chemotactic agents, including C3a and C4a, are produced.169 Mast cells degranulate under the influence of the latter, or IgE, and release ECF-A, NMW-NCF, ESM, histamine, and enzymes.170 Eosinophils and neutrophils, so recruited, bind (possibly via C3b receptors) to the basement membrane region. By direct cytotoxic action (eosinophils are capable of antibody-dependent cellular cytotoxicity) or via released proteases, particularly elastase, damage at the basement membrane region results in the development of a vesicle. Lymphocytes elaborate histamine-releasing factor (HRF), which increases mast cell degranulation and perpetuates the process. A broad range of cytokines are involved in this inflammatory reaction including interleukin (IL)-1, IL-4-IL-8, IL-10-IL-13, IL-15 and interferon gamma (IFN-γ).171 As yet, their relative importance and time sequences are unknown.
COOH
Lamina densa
react with BP180 that are readily detected by immunoblotting.142 However, the sera in 100% of patients react with BP180 NC16A domain recombinant protein.142 This latter finding underscores the usefulness of testing for anti-NC16A domain antibodies from peripheral blood to distinguish bullous pemphigoid from other disorders.111,143–145
Bullous pemphigoid is therefore a true autoimmune disease in which antigen–antibody reaction and complement fixation results in a characteristic and reproducible train of events, which is inevitably accompanied by the development of subepidermal blister formation. The etiology or initiator (other than those associated with drugs or PUVA therapy, which are the minority) is unknown. The question as to why self-tolerance breaks down with the formation of symptomatic autoantibodies in patients with this disease is an important question for further investigation.
Differential diagnosis The inflammatory cell-rich variant of bullous pemphigoid must be distinguished from other subepidermal blistering dermatoses in which a heavy inflammatory cell component is a typical finding. These include dermatitis herpetiformis, linear IgA disease, inflammatory epidermolysis bullosa
145 Pemphigoid gestationis
Parameter BP EBA BSLE LAD DHD
IMF Linear IgG, C3 Linear IgG, C3 Linear IgG, C3 Linear IgA Granular IgA
IIMF IgG antibodies 75–80% IgG antibodies 25–50% IgG antibodies 60% IgA antibodies 30% Antitransglutaminase antibodies
Split skin IMF Roof Floor Floor Roof or floor or both N/A
Type IV collagen Floor Roof Roof Roof or floor N/A
EM: site of split LL Sub-LD Sub-LD LL, sub-LD or both Papillary dermis
Western blot BP180 kD BP230 kD
290 kD (type VII collagen)
290 kD (type VII collagen)
BP180 kD BP230 kD 200/280 kD 285 kD 250 kD 290 kD
Antigen uncertain
BP, Bullous pemphigoid; BSLE, bullous systemic lupus erythematosus; DH, dermatitis herpetiformis; DIMF, direct immunofluorescence; EBA, epidermolysis bullosa acquisita; EM, electron microscopy; IIMF, indirect immunofluorescence; IMF, immunofluorescence; LAD, linear IgA disease; LL, lamina lucida; sub-LD, sub-lamina densa.
acquisita, and bullous systemic lupus erythematosus (see Table 4.6). Successful differentiation depends upon careful clinicopathologic correlation and immunofluorescent studies or, more recently, serum-based immunologic (ELISA) testing. Split skin indirect immunofluorescence or lamina densa antigen mapping by type IV collagen or laminin-1 direct immunoperoxidase is essential to determine the level of the split. Although electron microscopy, immunoelectron microscopy, and immunoprecipitation or Western blotting provide definitive information, such techniques are not necessary in the majority of cases.
where cases have had immunofluorescent confirmation would suggest that the latter figure is the most accurate.3
Pemphigoid gestationis may present in the first or any subsequent pregnancy.3 It may first also rarely present in the postpartum period. In one series, 30% of patients were primigravidae.9 In addition to developing in pregnant or postpartum patients, pemphigoid gestationis has rarely been described following a hydatidiform mole and gestational choriocarcinoma.11,12 It has not, however, been reported in nongestational variants, such as those occurring in the ovary, mediastinum, and testis, or complicating malignant teratoma. Pemphigoid gestationis is predominantly a disease of white females, being exceedingly rare in blacks.13,14 Presentation is usually in the second or third trimester, most often developing in the sixth or seventh month, but the range is variable from 2 months to 4 days postpartum.10,15 Although the disease may rarely completely remit before delivery, most patients (up to 75%) develop an exacerbation, which is frequently severe, in the immediate puerperium when progesterone levels have fallen.15,16 Exceptionally, the infant may show transient urticated erythema and blistering.4

Fig. 4.73 BP: direct immunoperoxidase reaction using frozen tissue substrate showing electron-dense deposits in the lamina lucida.

Fig. 4.74 BP: immunogold electron microscopic preparation. Note that the immunoreactant to BP180 and BP230 is particularly located on the hemidesmosomes (open arrows). However, deposits are also present within the lamina lucida, black arrows. (BC, basal cell; DER, dermis.) By courtesy of H. Shimizu, MD, Keio University School of Medicine, Tokyo, Japan.

Fig. 4.75 BP: Western blot demonstrating the two quite separate bullous pemphigoid antigens. By courtesy of M.M. Black, MD, Institute of Dermatology, London, UK.
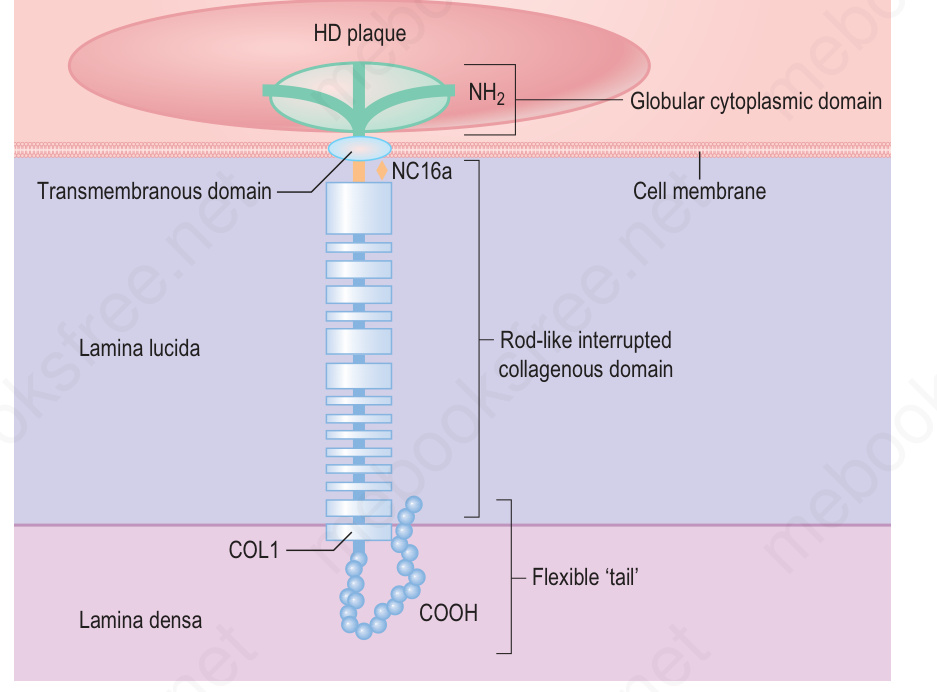
Fig. 4.76 A schematic representation of the BP180 molecule showing the globular intracellular NH2 domain, the membrane proximal NC16A domain and the flexible rod-like interrupted collagenous structure of the extracellular domain. (HD, hemidesmosome). Collagen XVII/BP180: a collagenous transmembrane protein and component of the dermal–epidermal anchoring complex. (Powell AM, Sakuma-Oyama Y, Oyama N, Black M.M. Department of Immunodermatology, St John’s Institute of Dermatology, St Thomas’ Hospital, London, UK.)
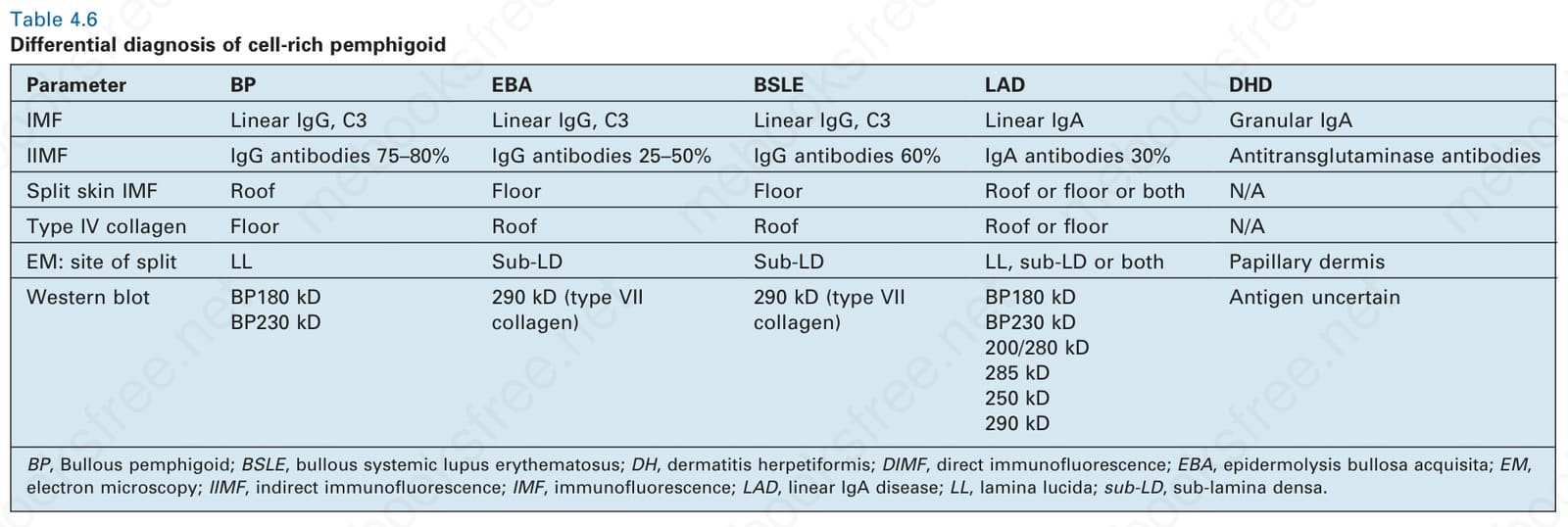
Table 4.6 Differential diagnosis of cell-rich pemphigoid
The cell-poor variant of bullous pemphigoid has a very wide range of differential diagnoses including epidermolysis bullosa (congenital and acquired), porphyria cutanea tarda, bullous amyloidosis, bullosa diabeticorum, and autolysis.