Olmsted syndrome
Olmsted syndrome
Clinical features Olmsted syndrome combines the features of mutilating palmoplantar keratoderma with periorificial plaques.1,2 The keratoderma is present at birth or begins in early infancy and when fully developed presents as bilateral and symmetrical massively thickened, yellow, macerated, keratotic plaques covering the whole of the soles and palms and often extending to the lateral and even the dorsal surface of the hands and feet (Fig. 3.104).3,4 The heels and forearms may also be affected. The border of the plaque is sharply defined and surrounded by a pruritic erythematous border. Lesions are often fissured and extremely painful, making walking exceedingly difficult or impossible.3,4 Blistering has occasionally been described.5 Flexion contractures, ainhum-like constriction bands, and autoamputation are common complications.
autosomal dominant trait.3 The PPK initially develops over pressure points, shows involvement of the back of the hands and increases with age. The keratoderma has a ‘pebbled’ appearance (Fig. 3.101) that may also be evident on the dorsal aspects of the digits, knees and elbows. Progressive hypotrichosis leading to alopecia affects the scalp, eyebrows, eyelashes, and axillary and genital regions. The nails, which may be normal at birth, gradually become thickened and dystrophic, displaying short nail plates that are easily shed (Fig. 3.102).4–6 The nail abnormalities may mimic those of pachyonychia congenita (PC) or other syndromes of ‘hair–nail hypoplasia’.7 Rare manifestations include sensorineural deafness, ocular abnormalities, skin hyperpigmentation, polydactyly, syndactyly, mental retardation, epilepsy, and dwarfism.5,6
Superinfection with bacteria and fungi, particularly Candida albicans, compounds the picture and as a result of this, lesions are frequently very malodorous. Development of squamous cell carcinoma or malignant melanoma has been reported.6–9
Affected children also develop erythematous keratotic papules and plaques around body orifices including the mouth, nostrils, ears, and anus.3,4 The eyelids, umbilical region, inguinal region, and gluteal cleft can also be involved. The keratotic lesions are pruritic and painful causing discomfort, particularly in the gluteal cleft.4,10,11
Additional features include scarring alopecia, keratosis pilaris, tooth anomalies, and nail dystrophy, including ridging, transverse striae,
95 Palmoplantar keratoderma
A
thickening, curvature, subungual keratosis, and infection. Hyperkeratotic linear streaks may develop in the axillae and cubital fossae. Growth retardation, laxity of the large joints, and corneal involvement are occasional manifestations.3,4,10,11
Pathogenesis and histologic features Olmsted syndrome may occur sporadically and in various inherited forms. In the sporadic, the autosomal dominant and in the autosomal recessive variants gain-of-function mutations in the transient receptor potential vanilloid 3 gene (TRPV3) could be detected.12,13 TRP cation selective ion channels are involved in hair growth, epidermal differentiation, and the modulation of inflammation, pain, and pruritus.14 TRPV3 mutations may also lead to elevated IgE levels with eosinophilia, erythromelalgia, and deafness.15 In the X-linked variant of Olmsted syndrome with alopecia universalis and severe nail dystrophy, specific mutations in the MBTPS2 gene have been identified. It encodes a zinc metalloprotease essential for cholesterol homeostasis, and endoplasic reticulum stress response.16 Interestingly, mutations in the same gene account for IFAP syndrome (ichthyosis follicularis with atrichia and photophobia), KFSD (keratosis follicularis spinulosa decalvans) and BRESEK/BRESHECK syndrome.
Histologically, the plaques are characterized by massive hyperkeratosis, often with foci of vertically orientated parakeratosis.2–5 There is hypergranulosis with large coarse granules under the former whereas the granular cell layer is absent beneath the areas of parakeratosis. The epidermis is acanthotic and shows psoriasiform hyperplasia or papillomatosis and there is edema and increased vascularity of the superficial dermis where a lymphohistiocytic infiltrate containing many mast cells is present.17–19
B
shows additional features, such as onychogryphosis, arachnodactyly, and acro-osteolysis.16,17

Fig. 3.101 Clouston syndrome: the keratoderma shows a typical ‘pebbled’ appearance.
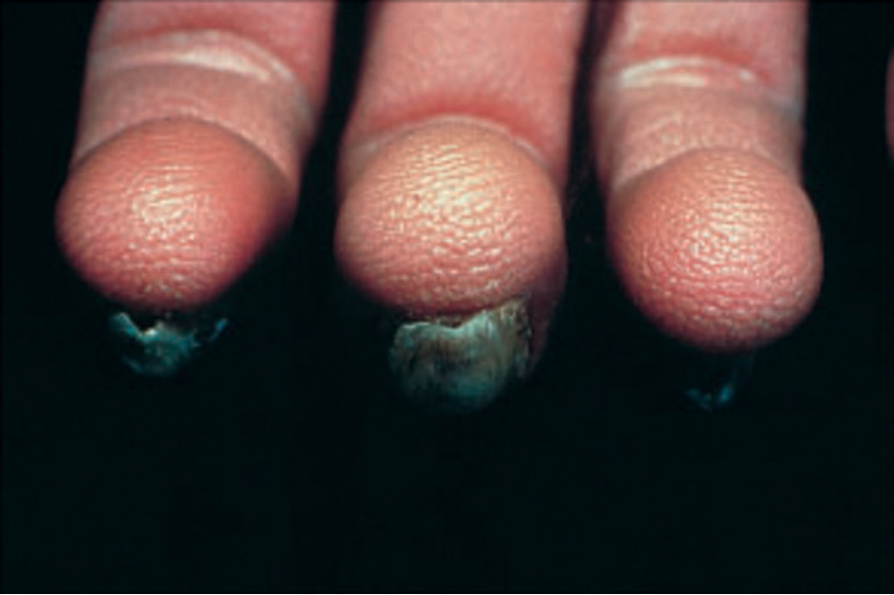
Fig. 3.102 Clouston syndrome: there is nail dystrophy accompanied by hyperkeratosis of the fingertips, thereby accentuating the epidermal surface ridges. By courtesy of D. Atherton, MD, the Children’s Hospital at Great Ormond Street, London, UK.
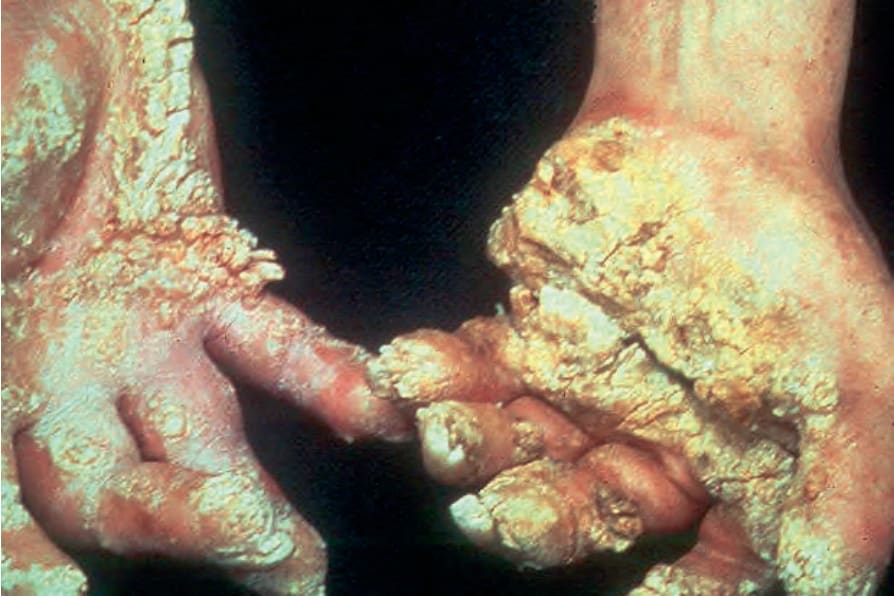
Fig. 3.104 Olmsted syndrome: in this variant, the lesions are very disfiguring. Constriction bands and autoamputation are important complications. By courtesy of W.A.D. Griffiths, MD, Institute of Dermatology, London, UK.
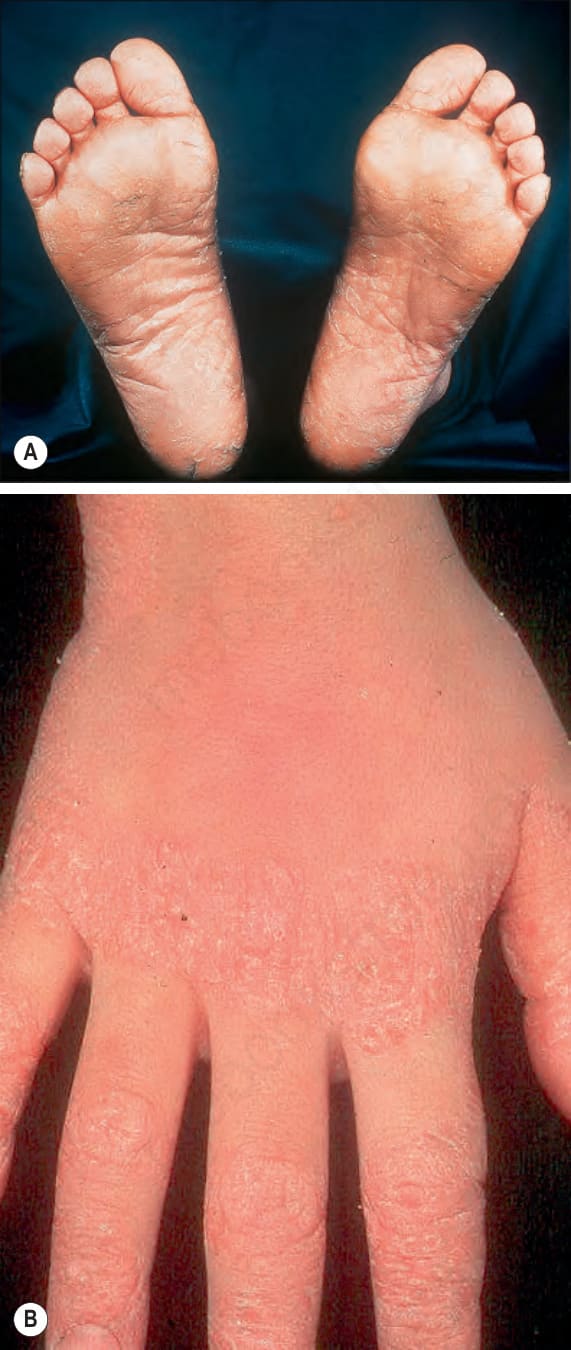
Fig. 3.105 Papillon-Lefèvre syndrome: (A) there is marked hyperkeratosis affecting the soles of the feet; (B) in this patient, the dorsal aspects of the hands, particularly the knuckles are also affected. By courtesy of W.A.D. Griffiths, MD, Institute of Dermatology, London, UK.