Peeling skin syndromes
Peeling skin syndromes
B
Peeling skin syndrome type B (inflammatory peeling skin disease) presents at birth with ichthyosiform erythroderma and is characterized by lifelong patchy peeling of the entire skin with severe pruritus. Isolated erythematous lesions show flaccid peeling, leaving burning superficially denuded red patches with a peripheral collarette (Fig. 3.46).5 Easy plucking of the hair and loose nail plates (onychomadesis) are further symptoms. Trigger factors are mechanical stress, low humidity or temperature changes. Pruritus and allergic symptoms of elevated IgE, food allergies, and asthma are reminiscent of Netherton syndrome.5 Autosomal recessive loss-of-function mutations of CDSN encoding corneodesmosin have been identified as the molecular cause of the disease.6
Clinical features Peeling skin syndromes (PSSs) are characterized by spontaneous peeling of the stratum corneum without bleeding or pain.1,2 Different genetic and clinical entities have to be distinguished.
In peeling skin syndrome type A (familial continual skin peeling, keratolysis exfoliativa congenitale) a generalized lifelong and continued shedding or peeling of the entire skin starts between 3–6 years of age.3 In contrast with the inflammatory type B, PSS type A lacks signs of inflammation or other symptoms or involvement of mucous membranes and nails. A missense mutation in CHST8 has been described that encodes a Golgi transmembrane N-acetylgalactosamine-4-O-sulfotransferase (GalNAc-4-ST1) that may be important for epidermal homeostasis resulting in increased and continuous desquamation of the stratum corneum.4
Histology shows a plane of separation either within the lower part of an otherwise normal horny layer or above the granular cell layer. Ultrastructural analysis reveals an intracellular splitting within the corneocytes.3
Histologically, the epidermis displays psoriasiform hyperplasia with an absent or reduced granular cell layer and marked parakeratosis. Careful examination reveals detachment of the horny layer from the granular cell layer (Fig. 3.47).5 Ultrastructurally, a loss of corneodesmosomes can be demonstrated, which is associated with intercellular splitting of the corneocytes from the stratum granulosum.6 Immunostaining for corneodesmosin and LEKTI may help to distinguish between Netherton syndrome and PSS type B.6
Acral peeling skin syndrome is characterized by superficial painless peeling of the backs of the hand and feet (Fig. 3.48). In children blistering occurs on palms and soles.7,8 Only recently it was reclassified as a form of
72 Disorders of keratinization
epidermolysis bullosa.9 Missense mutations in the gene of transglutaminase-5 (TGM5) represent the molecular cause of this autosomal recessive disease— in contrast to its major differential diagnosis epidermolysis bullosa simplex (EBS), which has an autosomal dominant inheritance.10 Other differential diagnoses of acral PSS include epidermolysis simplex superficialis9 and keratolysis exfoliativa.13,14 Histologically, the horny layer is detached from the stratum granulosum.7,8
Another type of PSS is related to the CSTA gene, mutations of which cause a deficiency of cystatin A. Initially, this variant was reported as ‘exfoliative ichthyosis’, a nonsyndromic congenital ichthyosis with dry and scaly skin associated with diffuse PPK sensitive to sweat and water exposure. The disease may display clinical features of SEI.11,12
Keratosis linearis-ichthyosis congenita-sclerosing keratoderma
Clinical features Keratosis linearis-ichthyosis congenita-sclerosing keratoderma (KLICK) is an autosomal recessive genodermatosis presenting with a moderate,
non-blistering ichthyosis from birth. Pathognomonic findings are keratotic punctuate plugs and papules arranged in parallel lines and circumscribed follicular keratosis around the wrists, in the folds of arms, axillae, and knees. In addition, a diffuse palmoplantar keratoderma develops including the dorsal side of hands and feet (Fig. 3.49). Other features include a sclerosing flexion deformity of the fingers and constriction bands.1,2 There are no other associated features, but there is a report of an associated squamous cell carcinoma at a younger age.3 Differential diagnosis includes loricrin keratoderma that also features palmoplantar keratoderma and a mild congenital ichthyosis but is less sclerotic and has an autosomal-dominant pattern on inheritance.
73 Ichthyosis
Pathogenesis and histologic features In KLICK a mutation of a proteasome maturation protein (POMP) has been identified. This protein serves as a chaperone for proteolytic enzymes degrading unneeded or damaged proteins. This proteasome insufficiency disturbs terminal epidermal differentiation and particularly interferes with processing of profilaggrin.4
Histologically, the epidermis is acanthotic and orthohyperkeratotic with focal parakeratosis and shows prominent hypergranulosis with irregular keratohyaline granules. This is reflected by immunohistochemistry for filaggrin which reveals extensive immunoreactivity in the upper epidermis that characteristically extends into the horny layer (Fig. 3.50). On non-volar skin follicular plugging is present. The papillary dermis shows papillomatosis and an inconsistent, mild perivascular lymphocytic infiltrate. Ultrastructure confirms the histologic finding of hypergranulosis and shows abnormally big keratohyaline granules.5
Severe dermatitis-multiple allergies-metabolic wasting syndrome (SAM)
This acronym was defined by Samuelov et al. in 2013 and refers to the cardinal features of severe dermatitis, multiple allergies, and metabolic wasting.1 SAM is another exfoliative disorder of cornification and thus resembles Netherton syndrome or PSS type B. SAM is caused by loss-offunction mutations in DSG1.1 Accordingly, apart from a psoriasiform dermatitis, subcorneal separation and acantholysis within the stratum spinosum and granulosum can be found. At the ultrastructural level half-split desmosomes are evident.1
umbilicus is said to be characteristic.5 Erythroderma is not a feature and the hair, nails, and sweat glands are unaffected.3,4 The diagnosis should be especially considered in preterm babies with congenital ichthyosis.5
The spasticity, which presents in early childhood, predominantly affects the legs and is often associated with contractures. The majority of patients are wheelchair bound.4 Kyphoscoliosis may also be present.3 Mental retardation is typically present, but is not invariable.1 Epilepsy occurs in up to 40% of patients.3
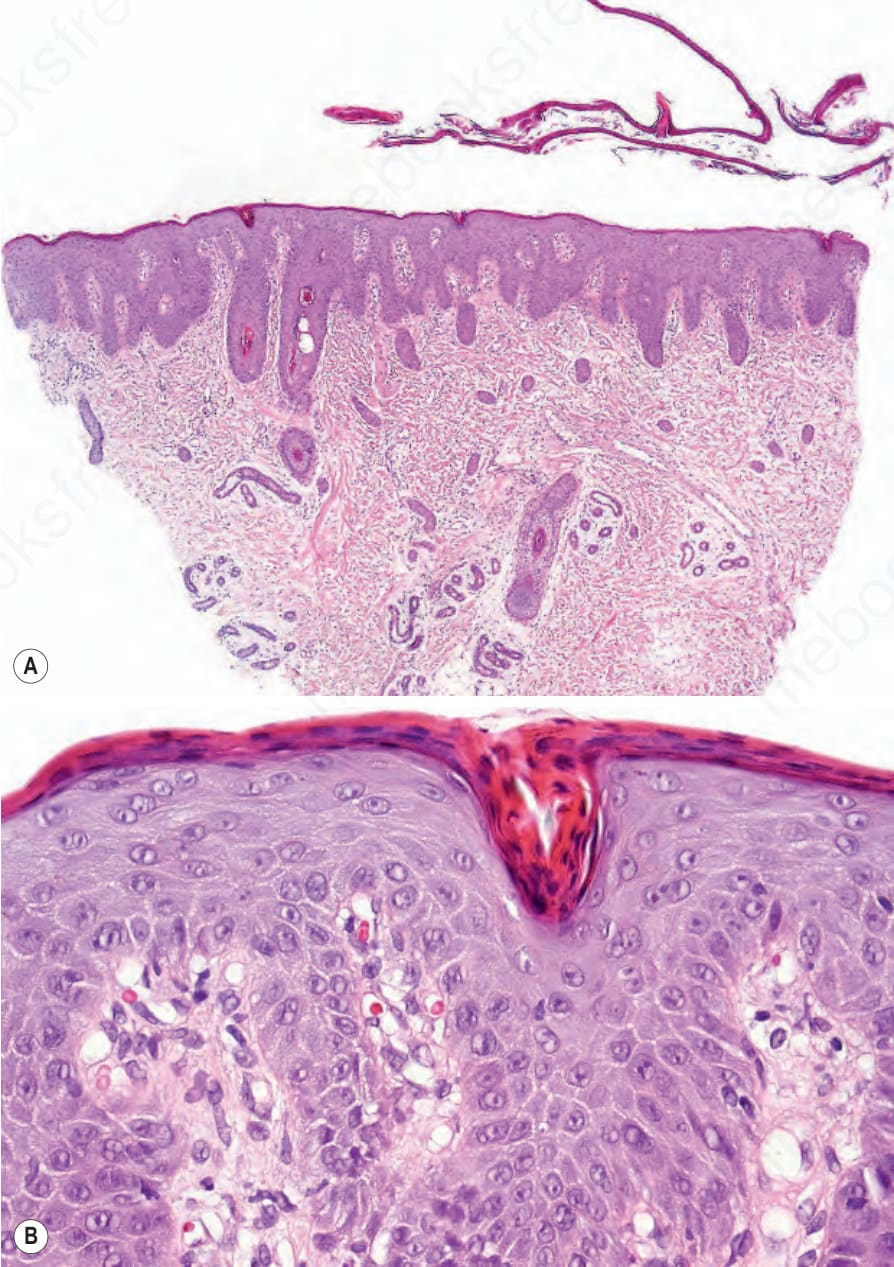
Fig. 3.45 Comèl-Netherton syndrome: (A) scanning view showing a detached thickened stratum corneum and psoriasiform hyperplasia; (B) note the marked parakeratosis and dilated vessels mimicking psoriasis vulgaris.
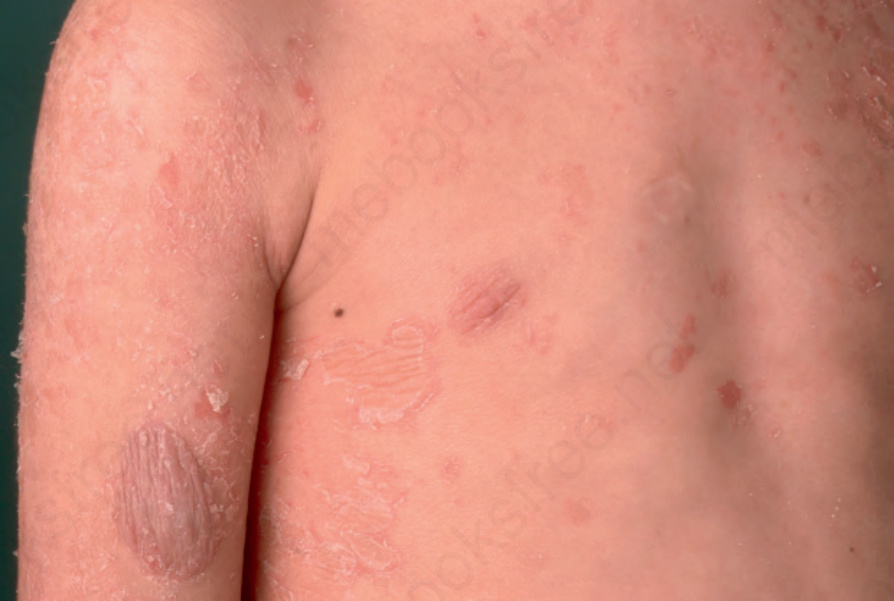
Fig. 3.46 Peeling skin syndrome type B: erythematous lesions show peeling of the skin leaving superficially denuded red patches. By courtesy of H. Traupe MD and V. Oji MD, Department of Dermatology, Münster, Germany.
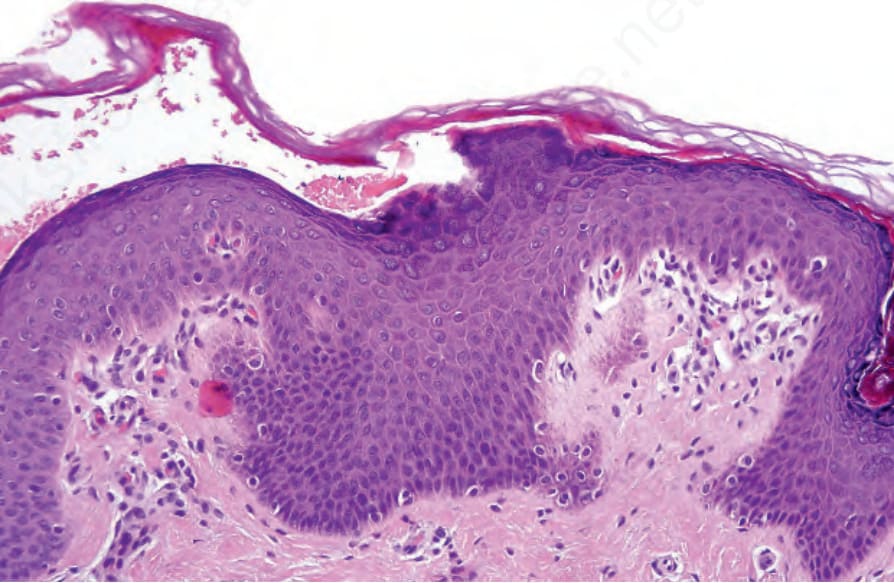
Fig. 3.47 Peeling skin syndrome type B: the biopsy is taken from the edge of the lesion. Note that the stratum corneum is clearly separated from the underlying epidermis.
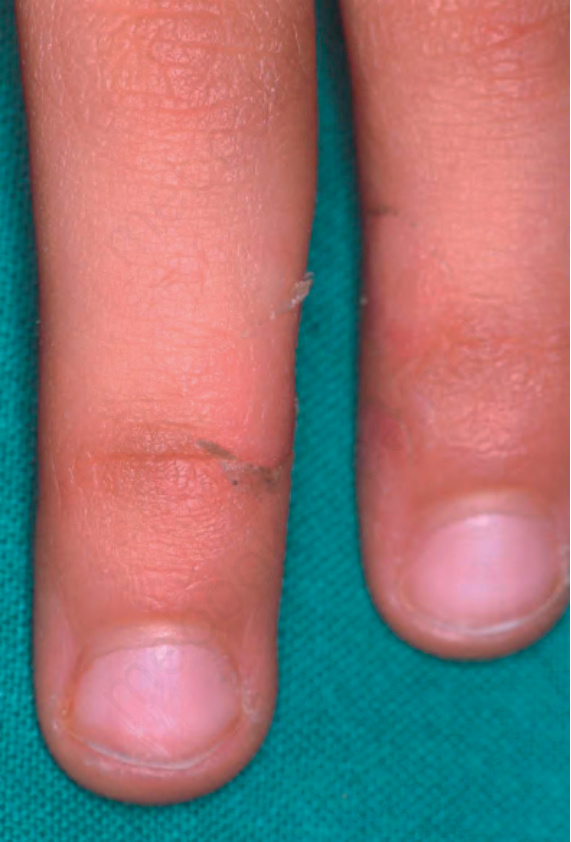
Fig. 3.48 Acral peeling skin syndrome type C: the skin of the backs of hand and feet shows reddish scaly patches. By courtesy of H. Traupe MD and V. Oji MD, Department of Dermatology, Münster, Germany.
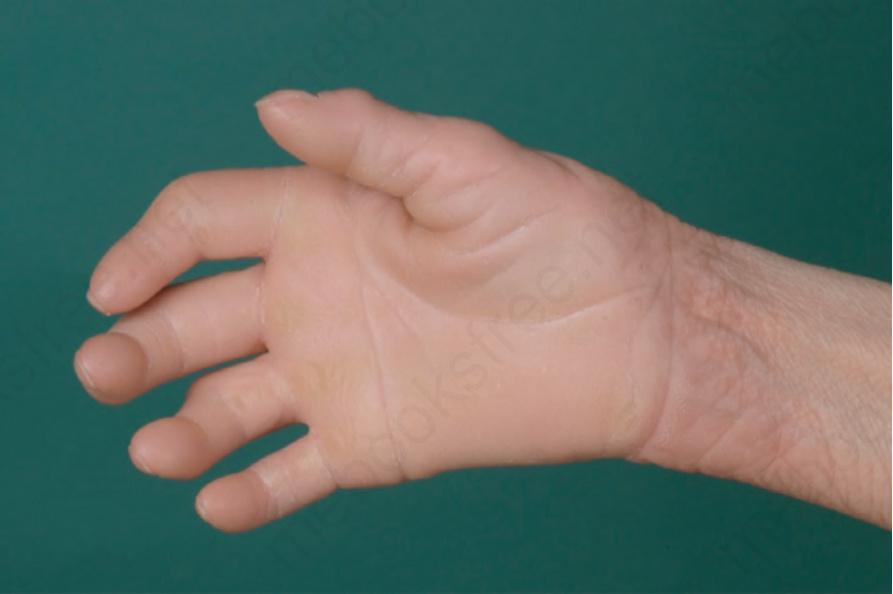
Fig. 3.49 Keratosis linearis-ichthyosis congenita-sclerosing keratoderma (KLICK): diffuse palmoplantar keratoderma and keratotic papules arranged in parallel lines on the wrist.
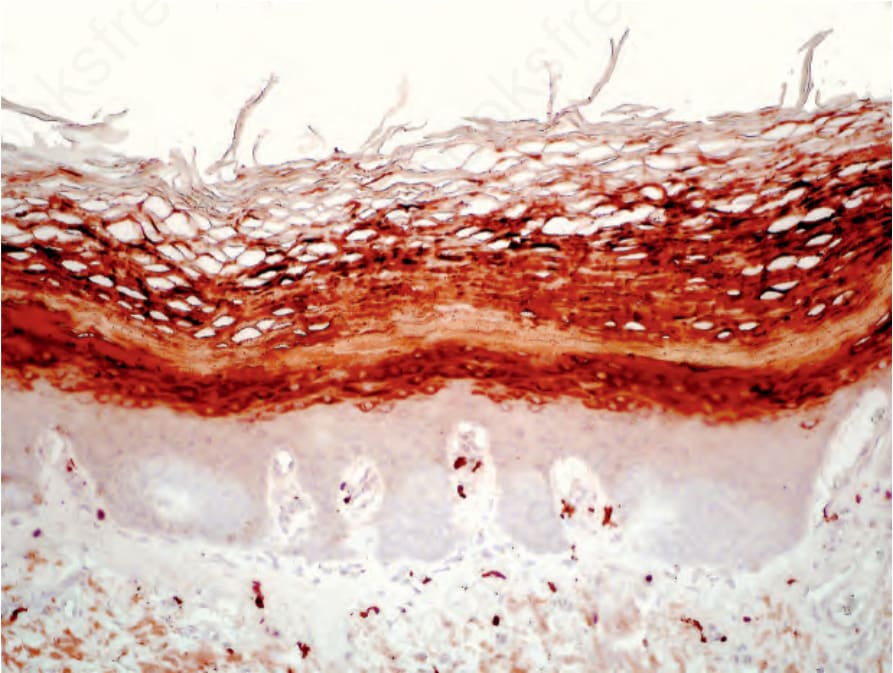
Fig. 3.50 Keratosis linearis-ichthyosis congenita-sclerosing keratoderma (KLICK): immunostaining for filaggrin shows broad immunoreactivity in the upper epidermis that characteristically extends into the horny layer.
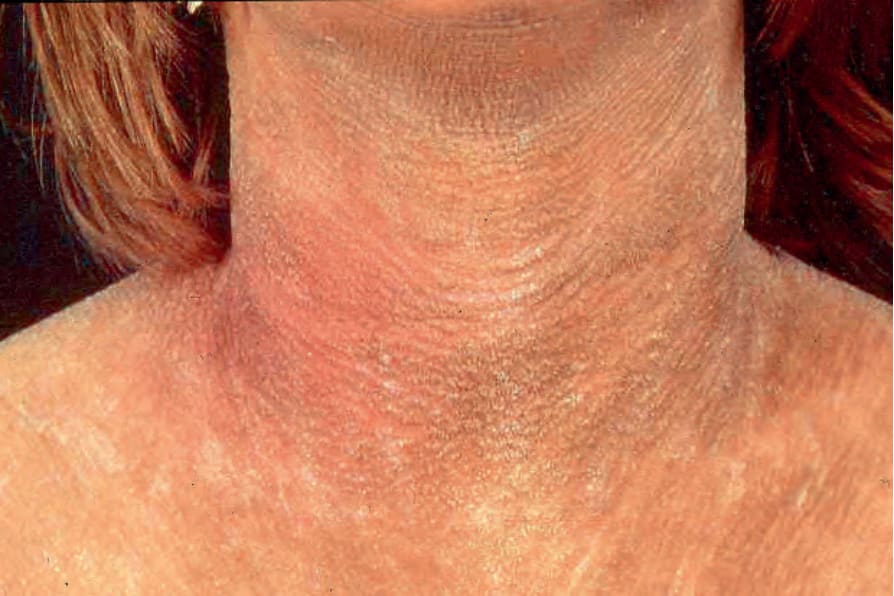
Fig. 3.51 Sjögren-Larsson syndrome: brownish-yellow color and a cobblestone-like pattern of lichenification is typical. By courtesy of M. Willemsen, MD, University Medical Center, Nijmegen, Belgium.
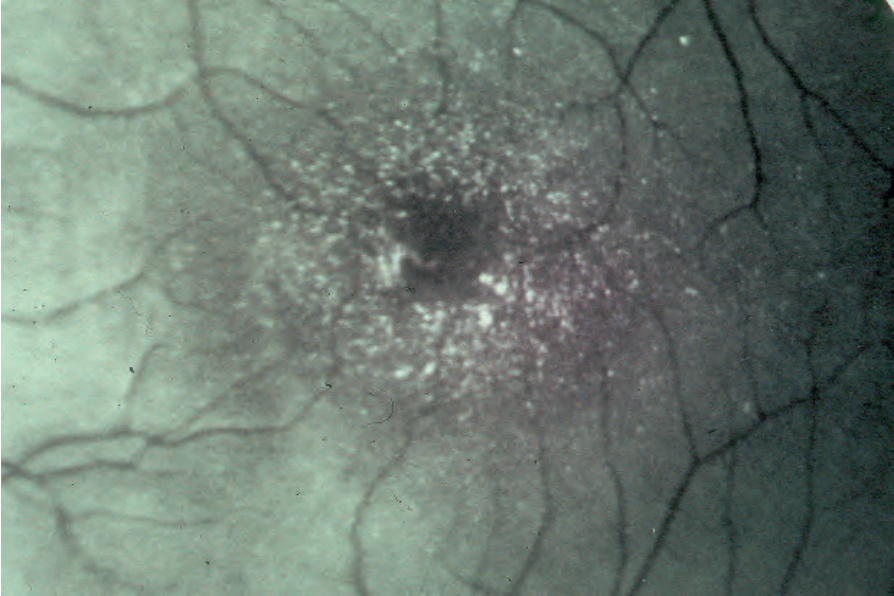
Fig. 3.52 Sjögren-Larsson syndrome: characteristic macular crystals. By courtesy of M. Willemsen, MD, University Medical Center, Nijmegen, Belgium.