Keratinopathic ichthyoses
Keratinopathic ichthyoses
According to the Sorèze Consensus conference in 2009, keratinopathic ichthyoses have been defined as a group of rare ichthyoses, which are caused by mutations in one of the keratin genes, namely keratin 1 (KRT1), keratin 10 (KRT10) or keratin 2 (KRT2). The old term ‘bullous congenital ichthyosiform erythroderma (of Brocq)’ has been renamed epidermolytic ichthyosis
63 Ichthyosis
A
B
diagnosis of this postzygotic mosaicism is mandatory as the patients risk developing full-blown keratinopathic ichthyosis.9,10
Differential diagnosis Epidermolytic hyperkeratosis is a histopathologic pattern that is seen in many conditions, including keratinopathic ichthyoses, palmoplantar keratoderma of the epidermolytic type epidermal nevus (see epidermolytic epidermal nevus), epidermolytic acanthoma, and epidermolytic leukoplakia (see Table 3.3). Epidermolytic hyperkeratosis has been shown to be one of the many patterns in Grover disease.11,12 It may also represent an incidental finding in normal, inflamed, and neoplastic skin (see incidental epidermolytic hyperkeratosis). Accurate clinical information is necessary to avoid diagnostic confusion.13
(EI); the term superficial epidermolytic ichthyosis (SEI) refers to the KRT2 associated subtype formerly known as ‘ichthyosis bullosa of Siemens’. Moreover, there are distinct keratinopathic ichthyosis forms, such as congenital reticular ichthyosiform erythroderma (CRIE) (also known as ichthyosis en confettis), ichthyosis Curth-Macklin, autosomal recessive EI, and annular EI (see Table 3.1).1
In the skin, basal keratinocytes predominantly express keratin 5 and 14 while suprabasal cells switch to the expression of keratin 1 and 10. Keratin 2e is only expressed in the subcorneal layer. Keratin monomers form obligate heterodimers in pairs of acidic (type I) and basic (type II) keratins, which assemble into keratin intermediate filaments building a cytoskeleton for the structural stability and flexibility of epidermal cells. Mutations in the higher molecular weight keratins 1 or 10 lead to collapse of the keratin skeleton in suprabasal keratinocytes while mutations in keratin 2 affect the upper layers. As a result the keratinocytes appear pale and show perinuclear shell formation or eosinophilic clumps, the latter changes known as epidermolytic hyperkeratosis.2–4 In these genodermatoses aggregation of the keratins is not permanent, but is inducible by pressure, high temperature, fever or skin infections. Epidermal sensitivity to hyperosmotic stress may be reduced by the chemical chaperone trimethylamine-N-oxide.5 Keratin aggregates also induce inflammation via interaction with the ubiquitin-proteasome system, activated MAP kinases, and chaperones, such as HSP70.7 Retinoids may interfere with collapse of the keratin skeleton in heat stressed keratinocytes with KRT10 mutation.6
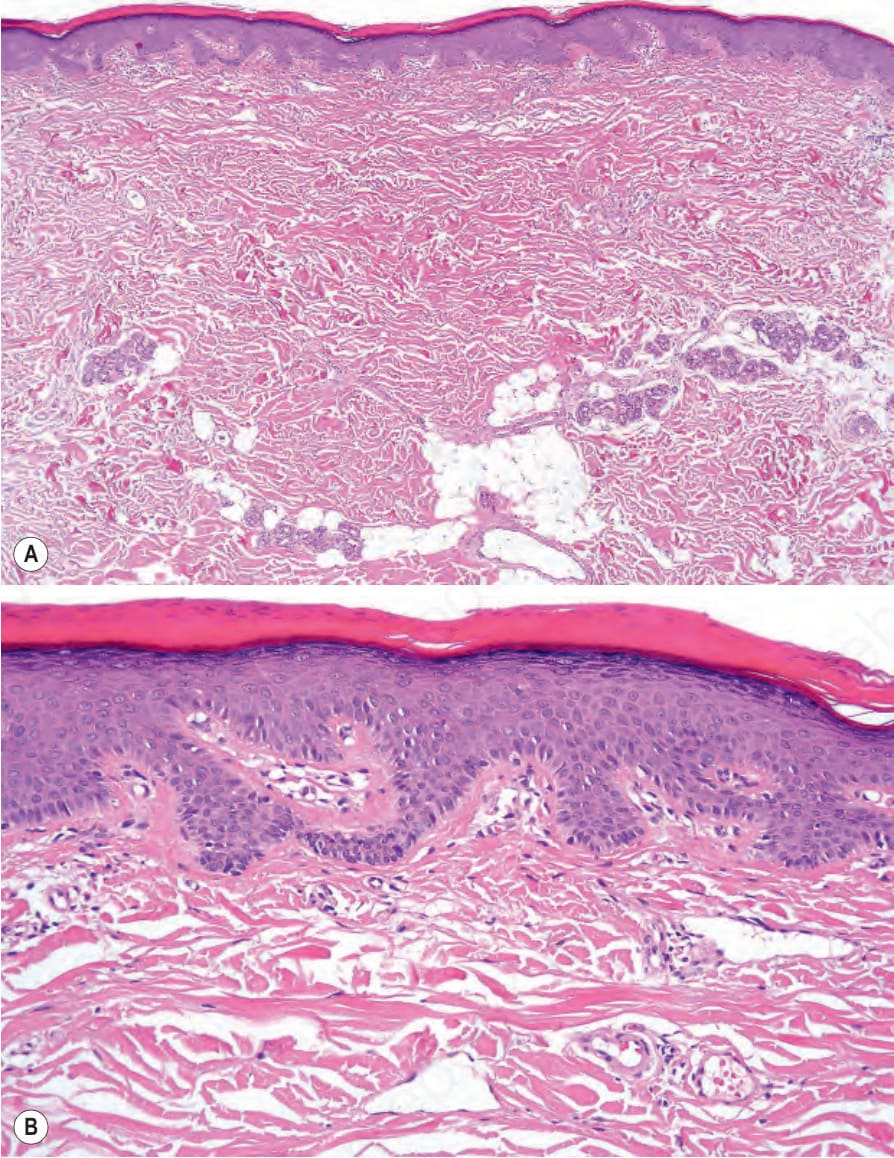
Fig. 3.22 (A, B) Autosomal dominant lamellar ichthyosis: there is a marked compact hyperkeratosis with focal parakeratosis and a prominent granular cell layer.
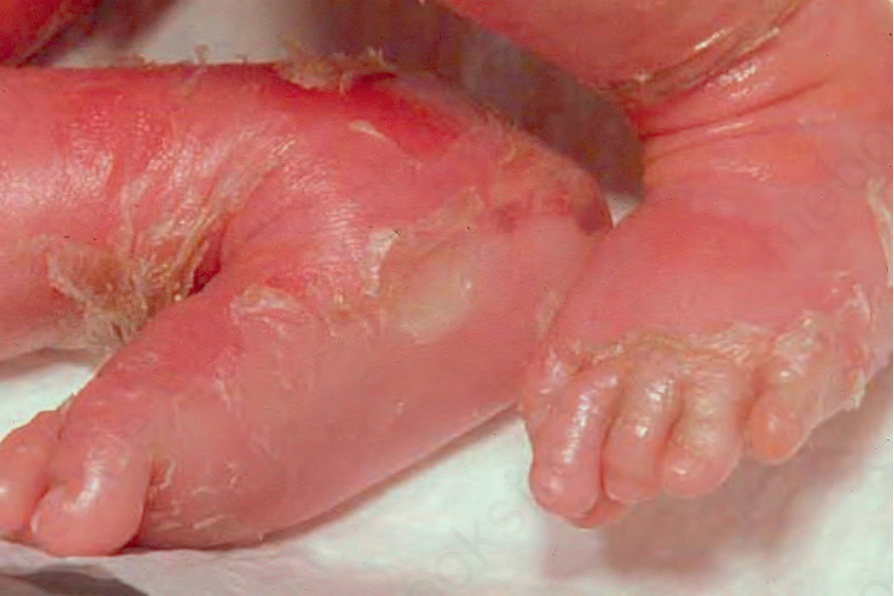
Fig. 3.23 Congenital bullous ichthyosiform erythroderma: close-up view of an infant showing intense erythema and blistering. By courtesy of M. Liang, MD, The Children’s Hospital, Boston, USA.
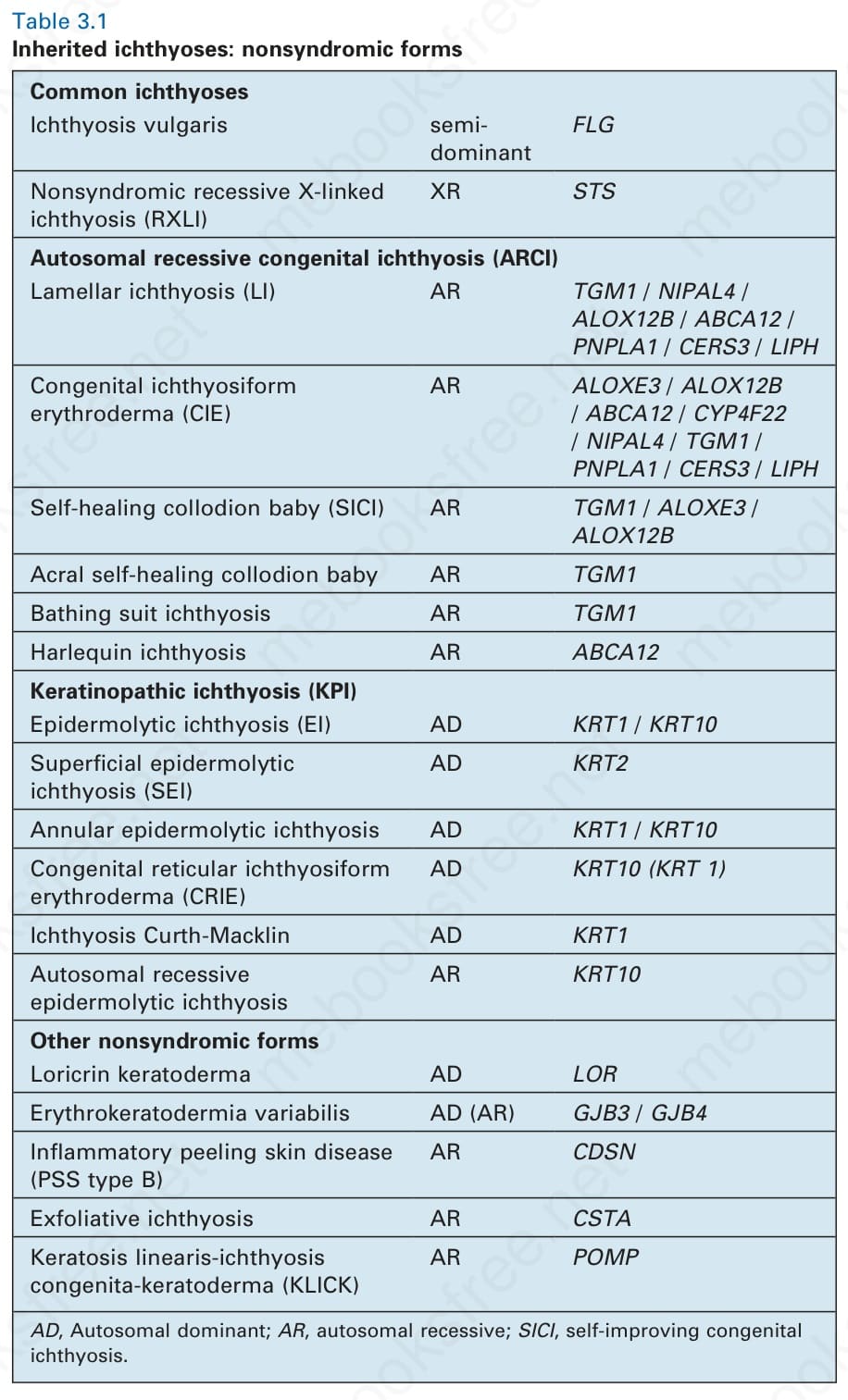
Table 3.1 Inherited ichthyoses: nonsyndromic forms
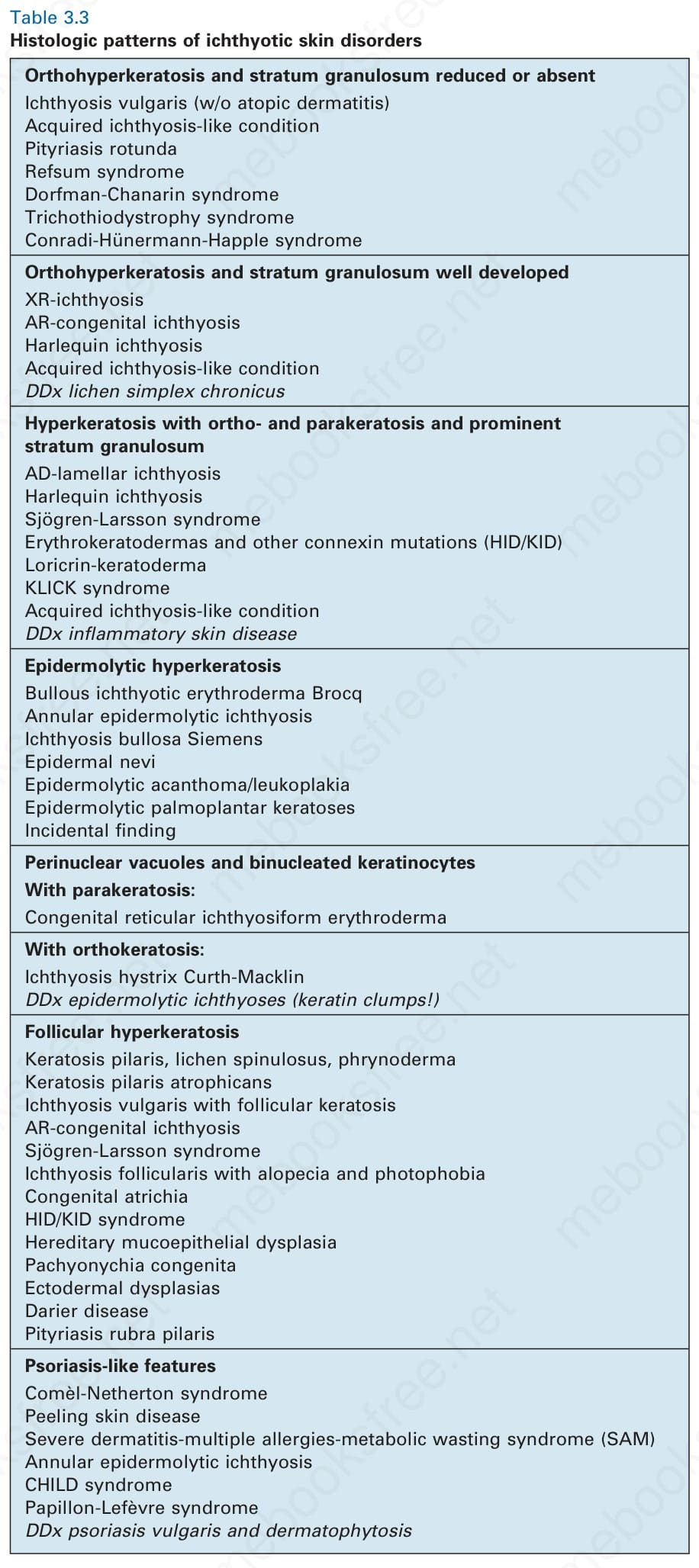
Table 3.3 Histologic patterns of ichthyotic skin disorders