X-linked (recessive) ichthyosis
X-linked (recessive) ichthyosis
Clinical features Also known as steroid sulfatase (STS) deficiency and ichthyosis nigricans, this X-linked, recessively inherited disorder has an incidence of 1 : 6000 male births.1–3 The disease is exceedingly rarely expressed in females.4 The disease may present at birth with dramatic but transient scaling of the skin, and this is sometimes misdiagnosed as autosomal recessive congenital ichthyosis. At the age of 2 to 6 months large and dark scales develop on the trunk, the extensor surface of the extremities, the scalp, the preauricular region, and the neck (Fig. 3.4).2 Mild involvement of the flexures is present (Fig. 3.5),1 but visible scaling may spare humid flexural regions of the skin.
56 Disorders of keratinization
Orthohyperkeratosis and stratum granulosum reduced or absent Ichthyosis vulgaris (w/o atopic dermatitis) Acquired ichthyosis-like condition Pityriasis rotunda Refsum syndrome Dorfman-Chanarin syndrome Trichothiodystrophy syndrome Conradi-Hünermann-Happle syndrome
A
Orthohyperkeratosis and stratum granulosum well developed XR-ichthyosis AR-congenital ichthyosis Harlequin ichthyosis Acquired ichthyosis-like condition DDx lichen simplex chronicus
Hyperkeratosis with ortho- and parakeratosis and prominent stratum granulosum AD-lamellar ichthyosis Harlequin ichthyosis Sjögren-Larsson syndrome Erythrokeratodermas and other connexin mutations (HID/KID) Loricrin-keratoderma KLICK syndrome Acquired ichthyosis-like condition DDx inflammatory skin disease
B
Epidermolytic hyperkeratosis Bullous ichthyotic erythroderma Brocq Annular epidermolytic ichthyosis Ichthyosis bullosa Siemens Epidermal nevi Epidermolytic acanthoma/leukoplakia Epidermolytic palmoplantar keratoses Incidental finding
Perinuclear vacuoles and binucleated keratinocytes With parakeratosis: Congenital reticular ichthyosiform erythroderma
Differentiation from ichthyosis vulgaris can be difficult as one-third of patients present with fine, light scales and the flexures may be spared in both diseases (Fig. 3.6). The palms and soles are usually unaffected and keratosis pilaris is not a feature, except if there is a concomitant deficiency of filaggrin and STS. Involvement of the trunk and neck often gives the skin a dirty appearance. Lesions may improve or disappear in warm weather.2 The hair, nails, and teeth are not affected.
Corneal opacities due to comma-shaped deposits in the posterior capsule of Descemet membrane or corneal stroma without visual impairment (Fig. 3.7), are characteristic and may be detected in female carriers.5 Inadequate cervical dilatation may lead to prolonged delivery of affected male newborns. Undescended testes and hypogonadism can be a feature in as many as 25% of affected patients.6–9 Rarely, testicular cancer has been documented.6 Interestingly, studies on STS deficiency have revealed an association with attention-deficit hyperactivity disorder (ADHD), and disorders within the spectrum of autism seem to occur more frequently.10
With orthokeratosis: Ichthyosis hystrix Curth-Macklin DDx epidermolytic ichthyoses (keratin clumps!)
Follicular hyperkeratosis Keratosis pilaris, lichen spinulosus, phrynoderma Keratosis pilaris atrophicans Ichthyosis vulgaris with follicular keratosis AR-congenital ichthyosis Sjögren-Larsson syndrome Ichthyosis follicularis with alopecia and photophobia Congenital atrichia HID/KID syndrome Hereditary mucoepithelial dysplasia Pachyonychia congenita Ectodermal dysplasias Darier disease Pityriasis rubra pilaris
Pathogenesis and histologic features The disease is associated with a deficiency of the microsomal enzyme steroid sulfatase/STS (sterol sulfate sulfohydrolase/arylsulphatase C).11 Absence of this enzyme is associated with persistence of the sulfate moiety on a number of sulfated steroid hormones and cholesterol sulfate.3
X-linked recessive ichthyosis is characterized by a raised serum cholesterol sulfate.12 The corneocytes contain excess cholesterol 3-sulfate and diminished free sterol,13 This may lead to persistence of the lipid contents
Psoriasis-like features Comèl-Netherton syndrome Peeling skin disease Severe dermatitis-multiple allergies-metabolic wasting syndrome (SAM) Annular epidermolytic ichthyosis CHILD syndrome Papillon-Lefèvre syndrome DDx psoriasis vulgaris and dermatophytosis
57 Ichthyosis
A
of the membrane-coating granules and hence increased or persistent adhesion between adjacent keratin plates in the stratum corneum. Beyond that, increased amounts of cholesterol sulfate may inhibit the epidermal serine protease activity, which results in retention of corneodesmosomes leading to less shedding of scales and retention hyperkeratosis.14 Steroid sulfatase deficiency can be detected using the patient’s peripheral leukocytes and cultured skin fibroblasts. Diagnosis may also be confirmed by lipoprotein electrophoresis, which shows increased mobility of low-density and very low-density beta-lipoproteins in addition to the steroid sulfatase deficiency.15,16
Technologies such as comparative genomic hybridization/comparative microarray analysis (CMA) allow rapid diagnosis in cases with large deletions.17 Carrier status can also be confirmed by fluorescent in situ
B
hybridization (FISH) analysis.18 Standard sequencing techniques are used to identify a point mutation that is the cause of the disease in approximately 10% of patients. Severity of this form of ichthyosis may be aggravated by a concomitant filaggrin mutation.19,20
Lesional skin shows compact orthohyperkeratosis and slight acanthosis associated with a granular cell layer, which may be normal or increased in thickness (Fig. 3.8).21,22 Keratohyalin granules show no abnormality.
58 Disorders of keratinization
A
B
Sjögren-Larsson syndrome and sometimes Netherton syndrome. Hence, collodion baby is a clinical description but not a disease. Still, the majority of these conditions develop into one of the ARCI subtypes, including SICI or SHCB.2,7
Follicular plugging is not a feature. Paradoxically, biopsies of thicker scales can show massive orthohyperkeratosis with reduction of the granular layer and a thin epidermis, causing confusion with ichthyosis vulgaris. A discrete lymphocytic perivascular inflammatory cell infiltrate may be evident.
Ultrastructural features include a high number of transitional cells and an abnormal persistence of desmosomal disks in the horny layer while keratohyalin granules are normal. An increased melanosome transfer accounts for the dark appearance of the scales.23
In the nonerythrodermic phenotype of congenital ichthyosis, the so-called ‘lamellar ichthyosis’, the scales are large, dark, and platelike, and cover the entire body, including the palms, soles, scalp, and flexures (Fig. 3.11).8–11 Fissuring of the hands and feet may occur and the skin around the joints may become verrucous. Many patients suffer from severe hypo- or anhidrosis with risk of hyperthermia, an important clinical symptom with great impact on the daily life of the patient.8 Nail dystrophy, hair involvement (scarring alopecia), severe ectropion (up to 80% of patients), and eclabium are characteristic (Fig. 3.12). The ectropion is of the cicatricial type and develops as a consequence of excessive dryness and associated contracture of the anterior lamella of the eyelid. Complications include corneal ulceration, vascularization, and corneal scarring with eventual blindness.11 Primary conjunctival lesions have been described including keratinization, hyper- and parakeratosis, and papilla development. The teeth are not affected.10
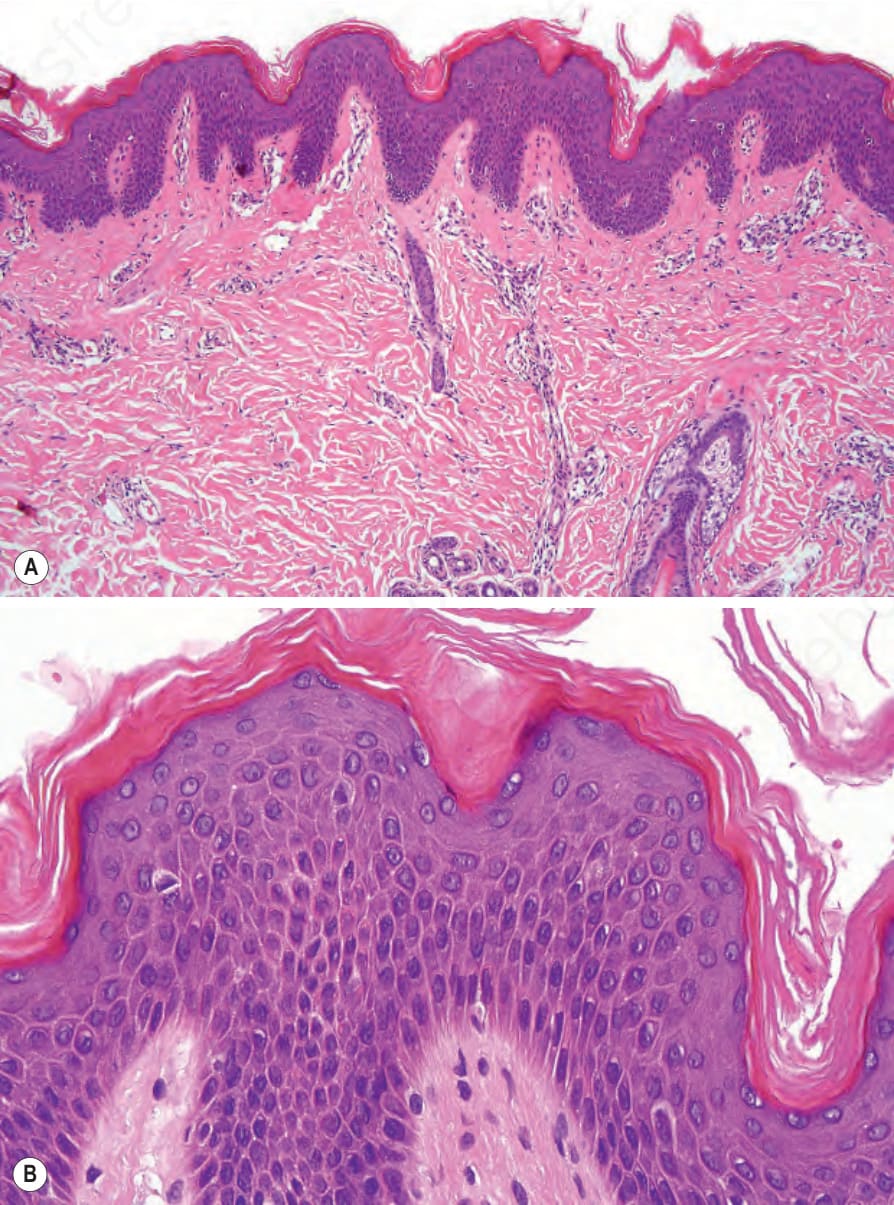
Fig. 3.3 (A, B) Ichthyosis vulgaris: there is orthohyperkeratosis with characteristic absence of the granular cell layer.
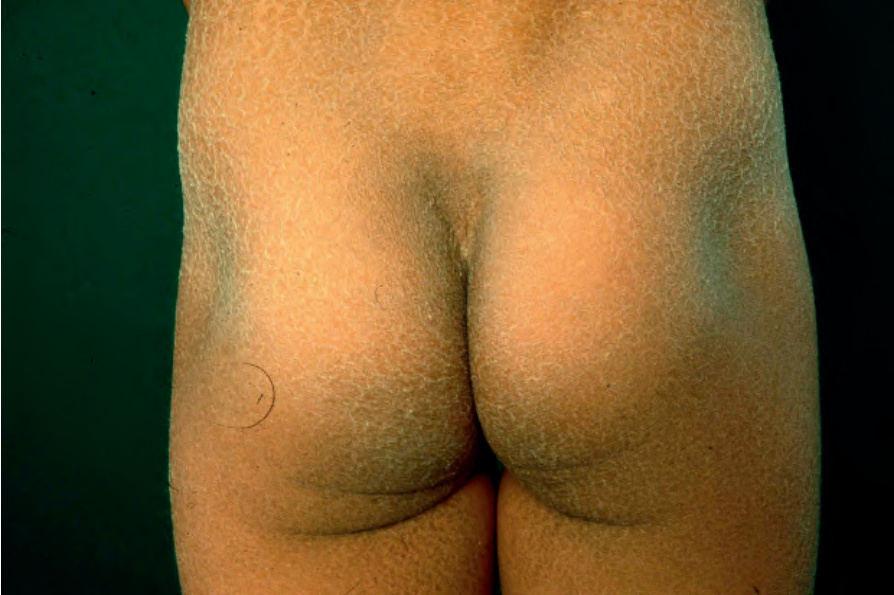
Fig. 3.4 X-linked recessive ichthyosis: many patients show large, confluent, and dark scales. By courtesy of the Institute of Dermatology, London, UK.
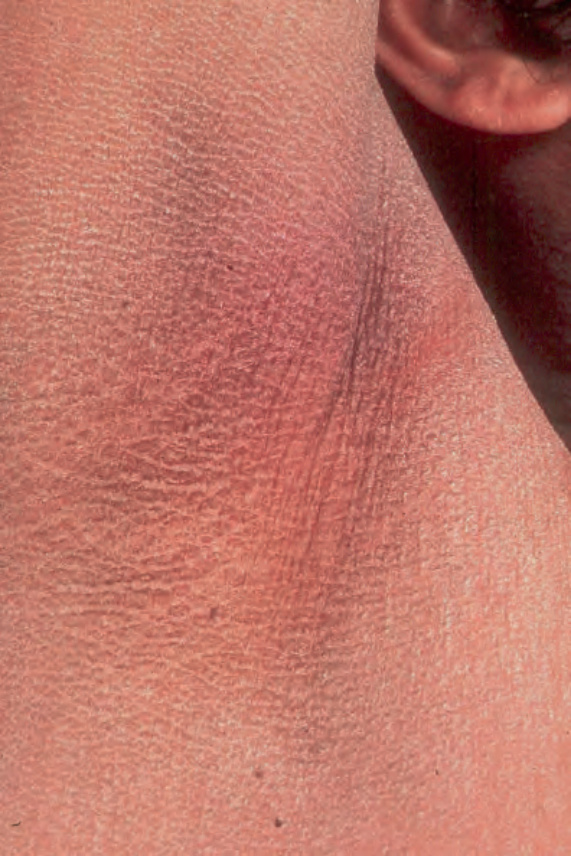
Fig. 3.5 X-linked ichthyosis: involvement of the flexures is a feature that allows differentiation form ichthyosis vulgaris. By courtesy of the Institute of Dermatology, London, UK.
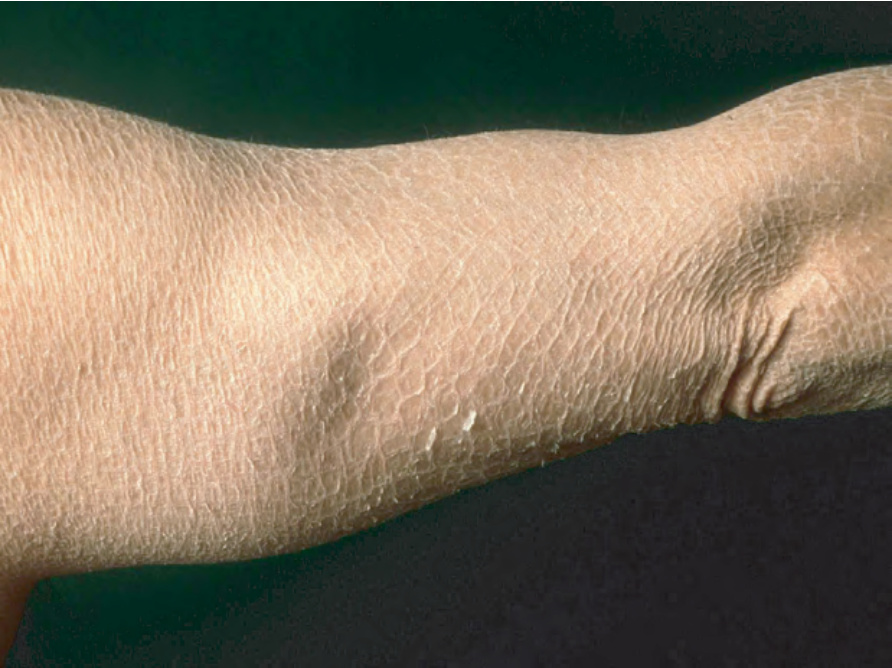
Fig. 3.6 X-linked recessive ichthyosis: some patients show light-gray scales.
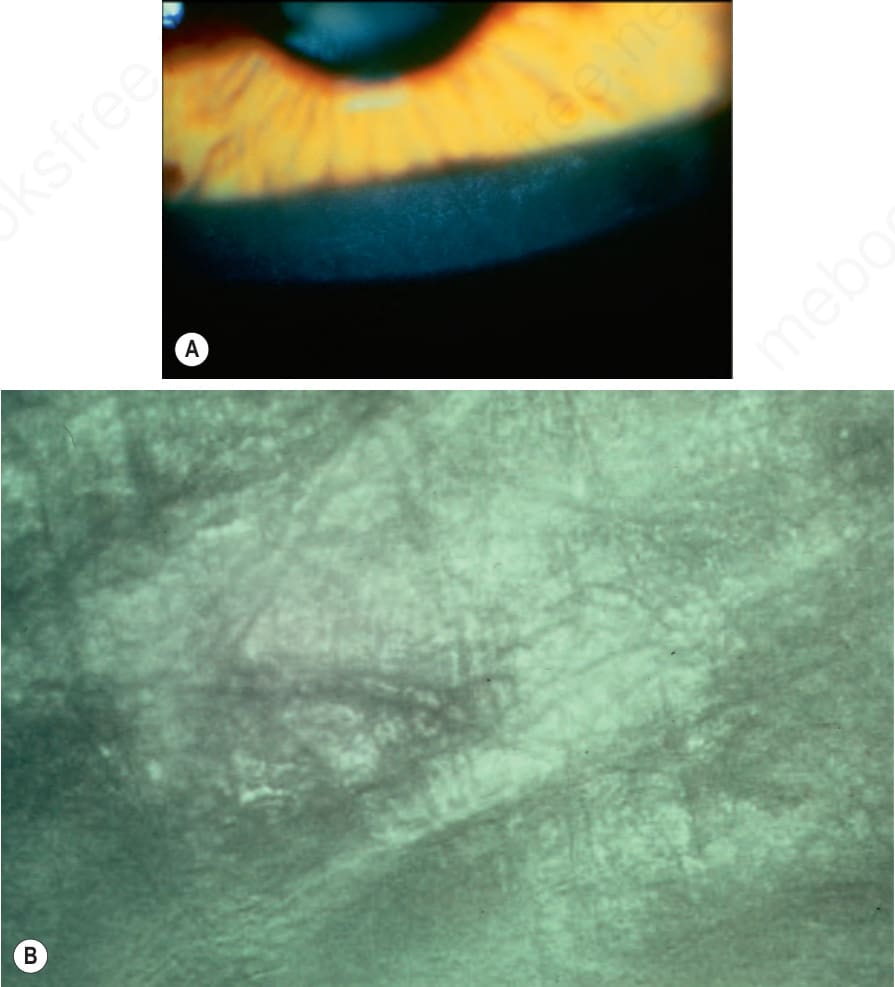
Fig. 3.7 (A) Sex-linked ichthyosis: characteristic linear opacities at the level of Descemet membrane. Slit-lamp photograph. (B) Same lesion viewed by specular microscopy. By courtesy of R.J. Buckley, MD, Moorfield’s Eye Hospital, London, UK.
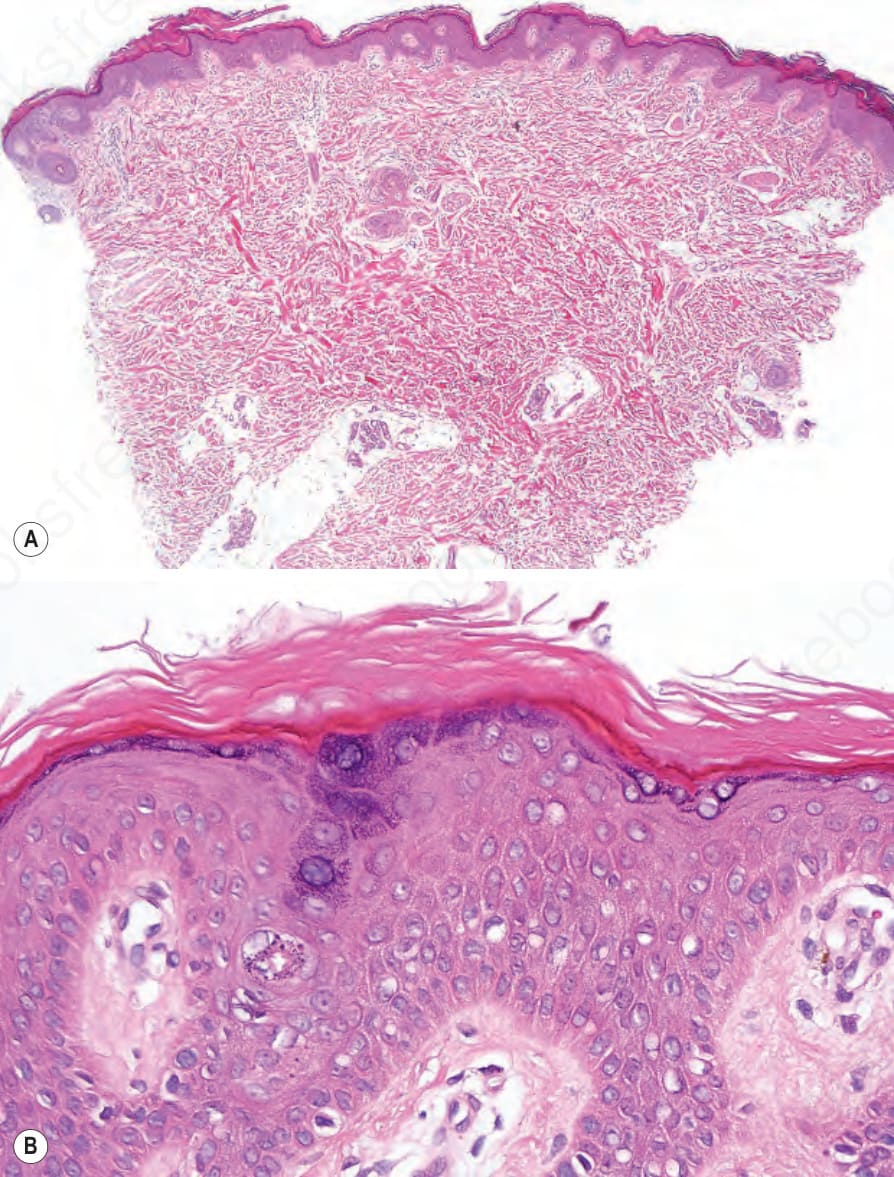
Fig. 3.8 (A, B) X-linked recessive ichthyosis: there is orthohyperkeratosis and mild acanthosis. The granular cell layer is normal.
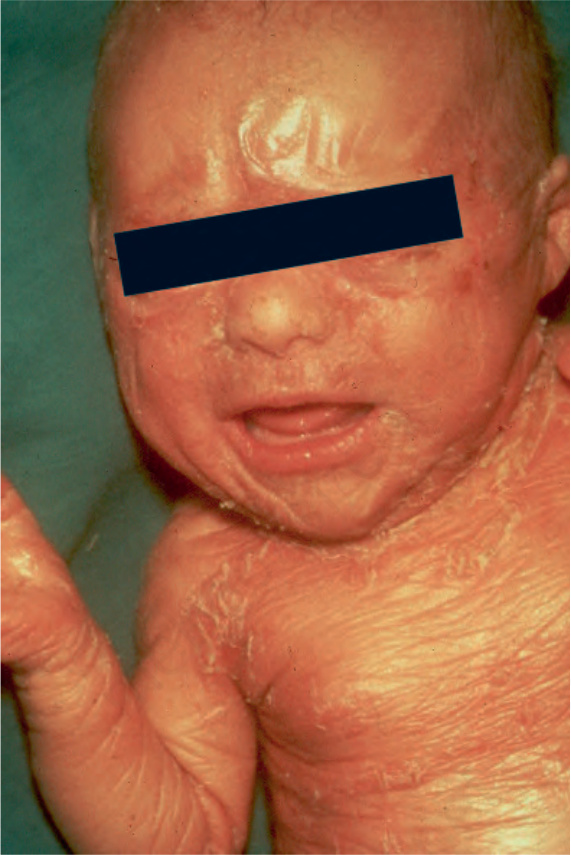
Fig. 3.9 Autosomal recessive congenital ichthyosis: the collodion membrane is best seen on the forehead. There is scaling and erythema on the trunk. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
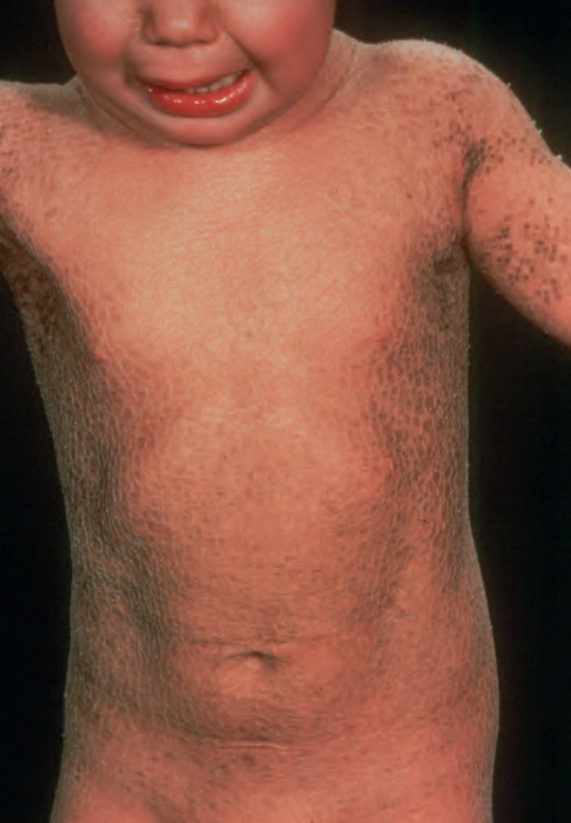
Fig. 3.11 Autosomal recessive lamellar ichthyosis: note the widespread and prominent large dark brown scales. By courtesy of D. Atherton, MD, Children’s Hospital at Great Ormond Street, London, UK.
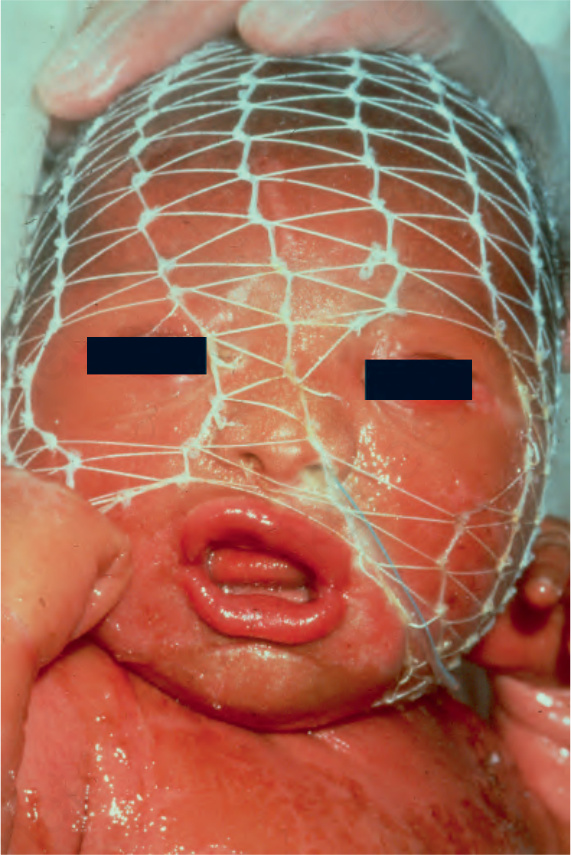
Fig. 3.12 Autosomal recessive lamellar ichthyosis: in this infant, there is gross ectropion and eclabion. By courtesy of D. Atherton, MD, Children’s Hospital at Great Ormond Street, London, UK.
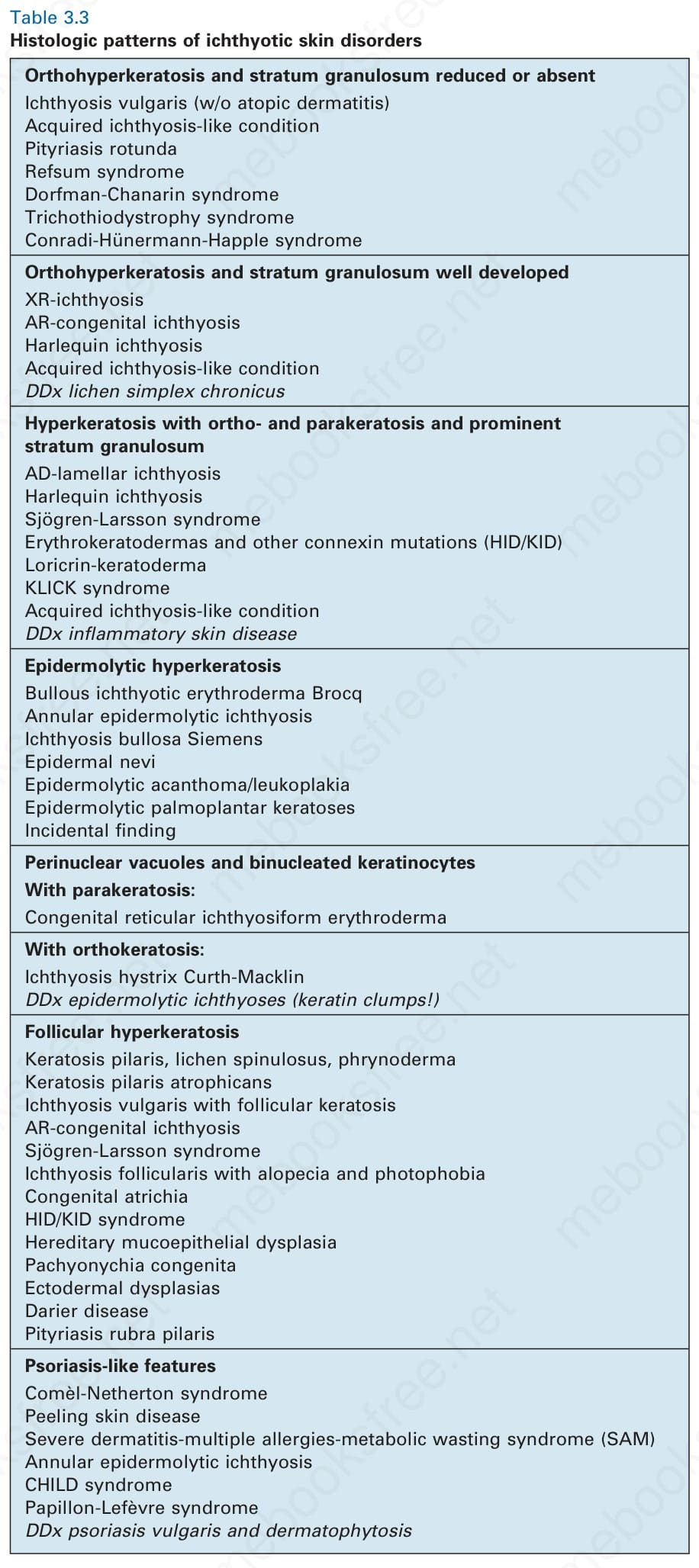
Table 3.3 Histologic patterns of ichthyotic skin disorders