Ichthyosis vulgaris
Ichthyosis vulgaris
Clinical features This relatively common disorder (incidence of 1 : 250 to 1 : 1000 births) has an autosomal semidominant mode of inheritance.1,2 The disease is usually fairly mild and becomes apparent within the first few months or years of life. It affects the sexes equally and presents as dryness (xerosis) and slight to moderate fine scaling, particularly involving the extensor surfaces of arms and legs and may spare the flexures (Fig. 3.2A). The light-gray scales vary in quality from thick adherent shiny plates to simply dusty accumulations which, when scratched, leave a mark just as when one touches a dusty
54 Disorders of keratinization
Common ichthyoses Ichthyosis vulgaris semidominant
FLG
Nonsyndromic recessive X-linked ichthyosis (RXLI)
XR STS
Autosomal recessive congenital ichthyosis (ARCI) Lamellar ichthyosis (LI) AR TGM1 / NIPAL4 / ALOX12B / ABCA12 / PNPLA1 / CERS3 / LIPH
Congenital ichthyosiform erythroderma (CIE)
AR ALOXE3 / ALOX12B / ABCA12 / CYP4F22 / NIPAL4 / TGM1 / PNPLA1 / CERS3 / LIPH
Self-healing collodion baby (SICI) AR TGM1 / ALOXE3 / ALOX12B
Acral self-healing collodion baby AR TGM1
Bathing suit ichthyosis AR TGM1
Harlequin ichthyosis AR ABCA12
Keratinopathic ichthyosis (KPI) Epidermolytic ichthyosis (EI) AD KRT1 / KRT10
A
Superficial epidermolytic ichthyosis (SEI)
AD KRT2
Annular epidermolytic ichthyosis AD KRT1 / KRT10
Congenital reticular ichthyosiform erythroderma (CRIE)
AD KRT10 (KRT 1)
Ichthyosis Curth-Macklin AD KRT1
Autosomal recessive epidermolytic ichthyosis
AR KRT10
Other nonsyndromic forms Loricrin keratoderma AD LOR
Erythrokeratodermia variabilis AD (AR) GJB3 / GJB4
Inflammatory peeling skin disease (PSS type B)
AR CDSN
Exfoliative ichthyosis AR CSTA
Keratosis linearis-ichthyosis congenita-keratoderma (KLICK)
AR POMP
AD, Autosomal dominant; AR, autosomal recessive; SICI, self-improving congenital ichthyosis.
Pathogenesis Ichthyosis vulgaris is caused by loss-of-function mutations of the FLG gene that encodes for filaggrin, the major constituent of the keratohyalin granules.5,6 Patients may have one or two FLG mutations. The mutation status correlates with disease severity and ultrastructure, i.e., in line with the decreased amount of filaggrin, keratohyalin granules may be reduced, spongy or slightly crumbly.7 Filaggrin aggregates keratin intermediate filaments in the lower stratum corneum and is subsequently proteolyzed to form free amino acids, including urocanic and pyrrolidone carboxylic acids, which are critical as water-binding compounds in the stratum corneum. The clinical severity of ichthyosis vulgaris correlates with the reduction of keratohyalin granules. Reduced immunostaining for filaggrin correlates with the severity of the defect.8 Parents with one heterozygous filaggrin mutation may be asymptomatic, whereas affected offspring with two mutations often show classic ichthyosis vulgaris.5 FLG mutations are a major predisposing factor for atopic dermatitis (AD) and related allergies.9,10
B
surface. The truncal lesions tend to be thicker than those on the face and scalp. The rims of the ears are often scaly.3 There is a mild seasonal variation, with improvement of the condition in humid climates.2 The palms and soles show increased palmar and plantar markings (hyperlinearity), in contrast to pure X-linked ichthyosis (Fig. 3.2B).3 Affected patients often present keratosis pilaris (follicular hyperkeratosis) on the arms, buttocks and thighs and suffer from hypohidrosis. An association with keratosis punctata of the palms and soles has also been documented.4
Histologic features Ichthyosis vulgaris is characterized by mild to moderate orthohyperkeratosis associated with an acanthotic, atrophic or normal epidermis. The key
Mode of inheritance Gene
X-linked ichthyosis syndromes Recessive X-linked ichthyosis XR STS (and others)
Ichthyosis follicularis alopecia photophobia
XR MBTPS2
55 Ichthyosis
Conradi-Hünermann-Happle syndrome (CDPX2)
XD EBP
Autosomal ichthyosis syndromes with prominent hair abnormalities Netherton syndrome AR SPINK5
Severe dermatitis-multiple allergiesmetabolic wasting (SAM)
AR DSG1
Ichthyosis with hypotrichosis AR ST14
Neonatal ichthyosis-sclerosing cholangitis (NISCH)
AR CLDN1
Autosomal ichthyosis syndromes with prominent neurologic signs Refsum syndrome AR PHYH / PEX7
Multiple sulfatase deficiency AR SUMF1
Gaucher syndrome type 2 AR GBA
Sjögren-Larsson syndrome AR ALDH3A2
Dorfman-Chanarin syndrome AR ABHD5
Trichothiodystrophy AR C7ORF11 ERCC2 / XPD ERCC3 / XPB GTF2H5 / TTDA
Cerebral dysgenesis-neuropathyichthyosis-palmoplantar keratoderma (CEDNIK)
AR SNAP29
Arthrogryposis-renal dysfunction-cholestasis
AR VPS33B
A
Autosomal ichthyosis syndromes with deafness Keratitis ichthyosis deafness (KID) AD GJB2 (GJB6)
ELOVL4 deficiency AR ELOVL4
Mental retardation-enteropathy-deafnessneuropathy-ichthyosis-keratoderma (MEDNIK)
AR AP1S1
Autosomal ichthyosis syndromes with transient neonatal respiratory distress Ichthyosis prematurity syndrome AR SLC27A4
AD, Autosomal dominant; AR, autosomal recessive; CDPX2, chondrodysplasia punctata type 2.
B
feature is a thin or absent granular cell layer (Fig. 3.3).11,12,13 Regional variation in the thickness and/or presence of the granular cell layer may occur and therefore it is best to take the biopsy from a site of maximal scaling. The lesions of keratosis pilaris show dilated follicles containing large keratin plugs. In the upper dermis a mild perivascular lymphocytic infiltrate may be present. When ichthyosis vulgaris is associated with atopic dermatitis, parakeratosis and other signs of a spongiotic dermatitis can be found.
Differential diagnosis The histologic differential diagnosis includes other diseases characterized by orthohyperkeratosis and a reduced or absent stratum granulosum (Table 3.3).
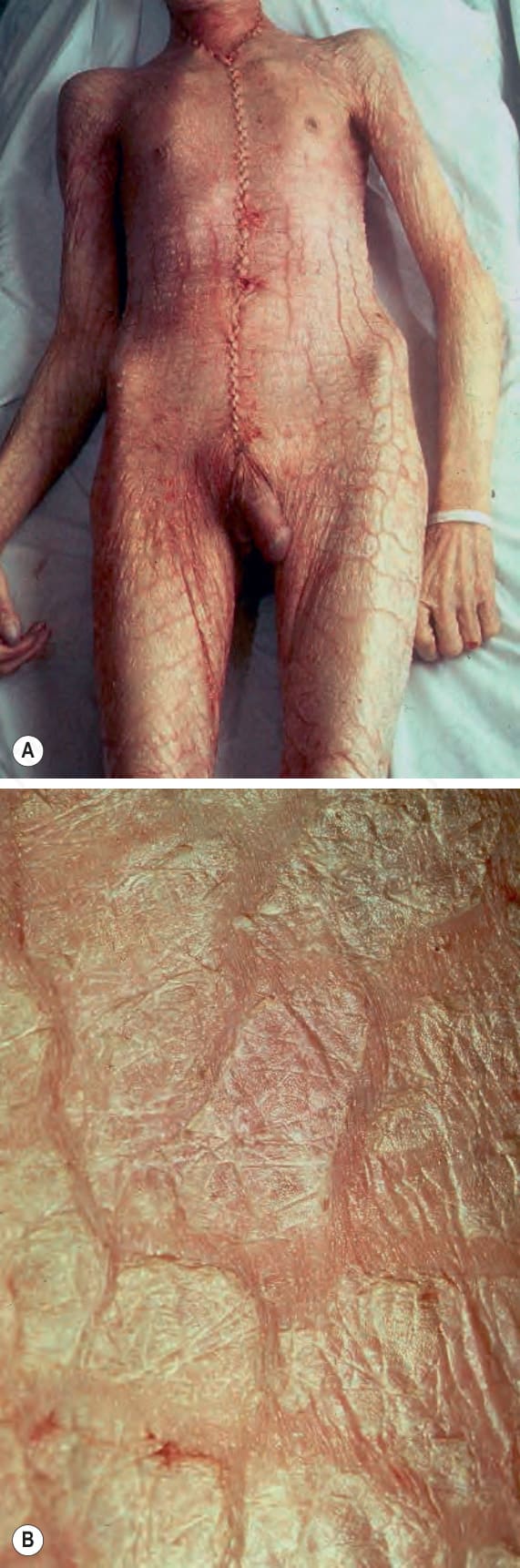
Fig. 3.1 (A, B) Severe generalized ichthyosis: this was an incidental finding at autopsy. Ichthyosis can be very disfiguring and a considerable social disadvantage. (A,B) By Courtesey Ph. McKee.
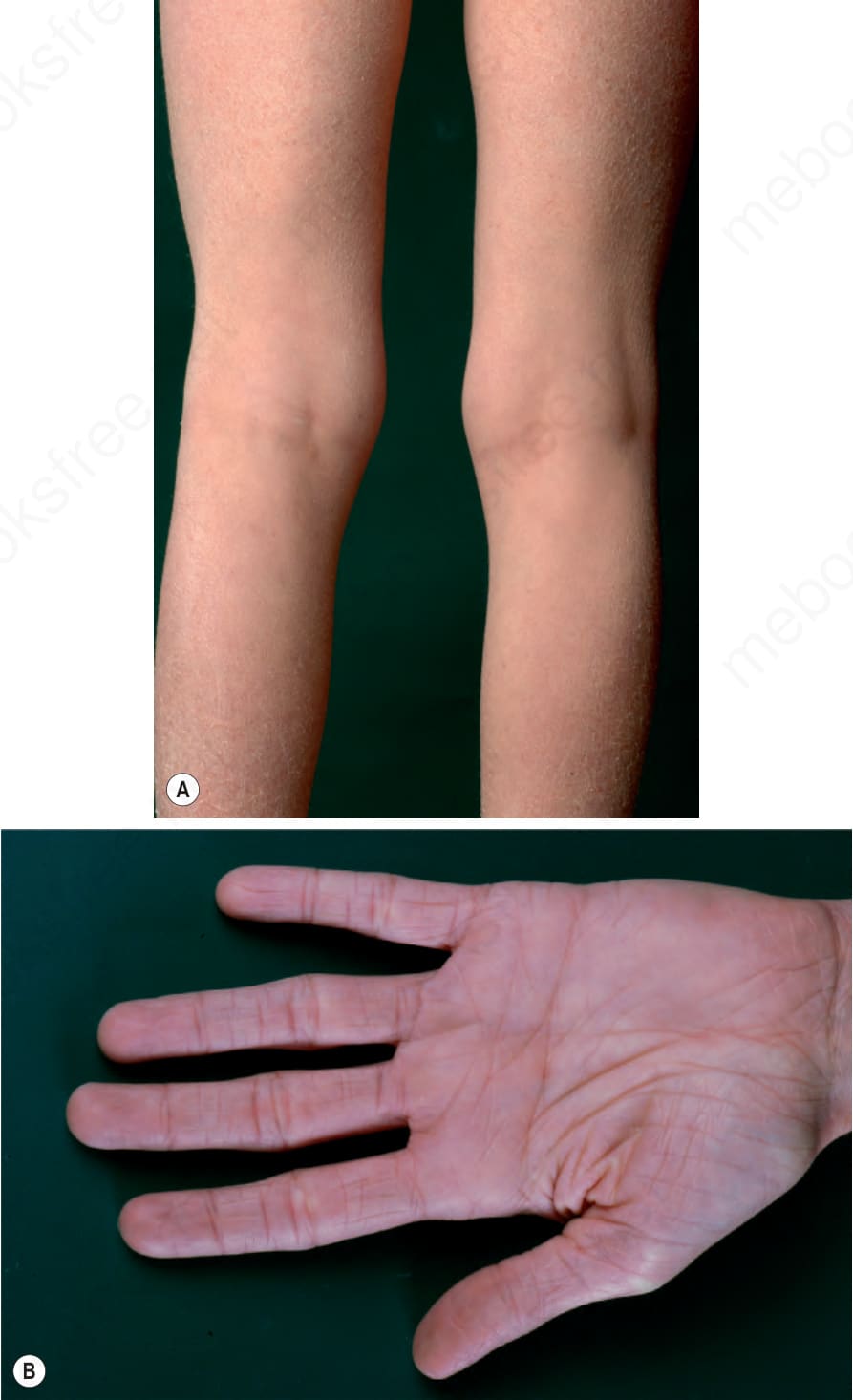
Fig. 3.2 (A) Ichthyosis vulgaris: fine scaling, particularly involving the extremities and characteristically sparing the flexures. (B) Palms show typically increased skin markings.
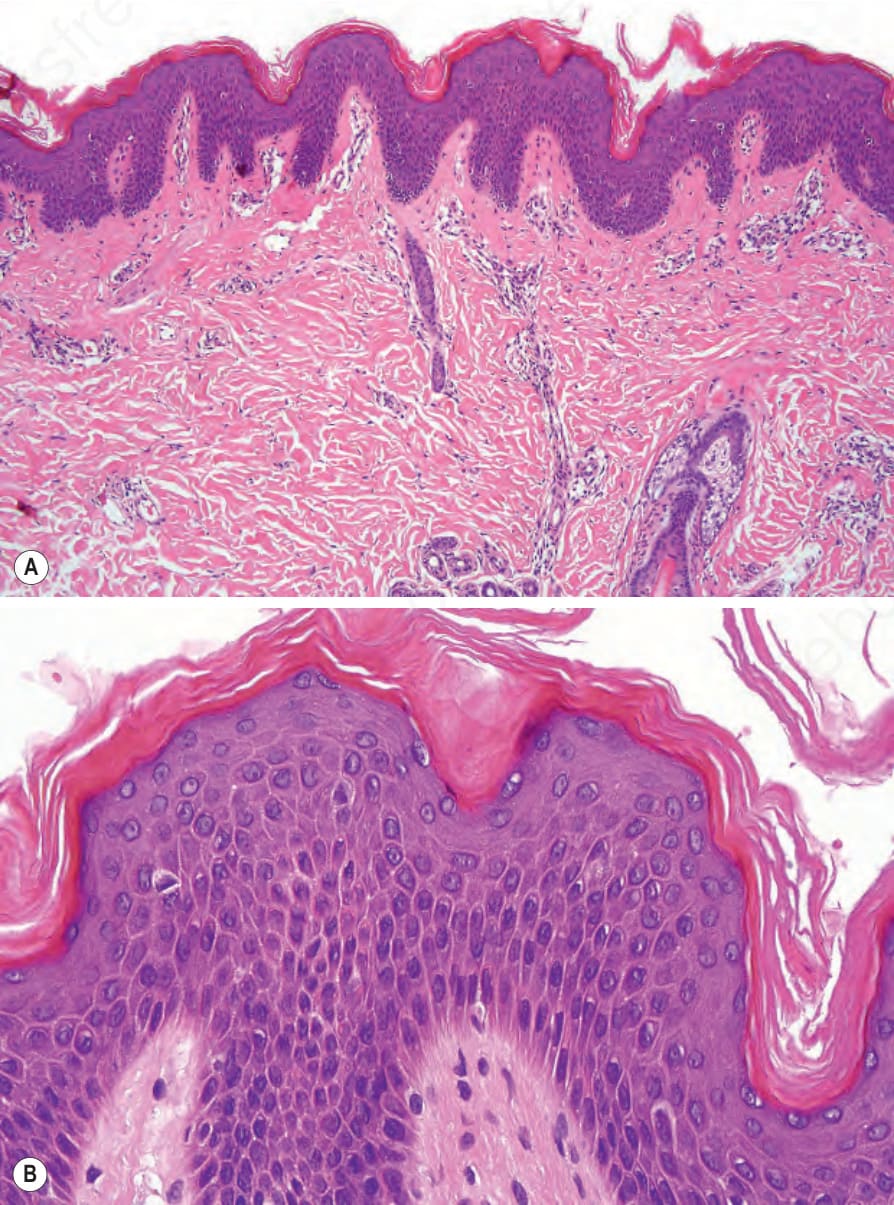
Fig. 3.3 (A, B) Ichthyosis vulgaris: there is orthohyperkeratosis with characteristic absence of the granular cell layer.
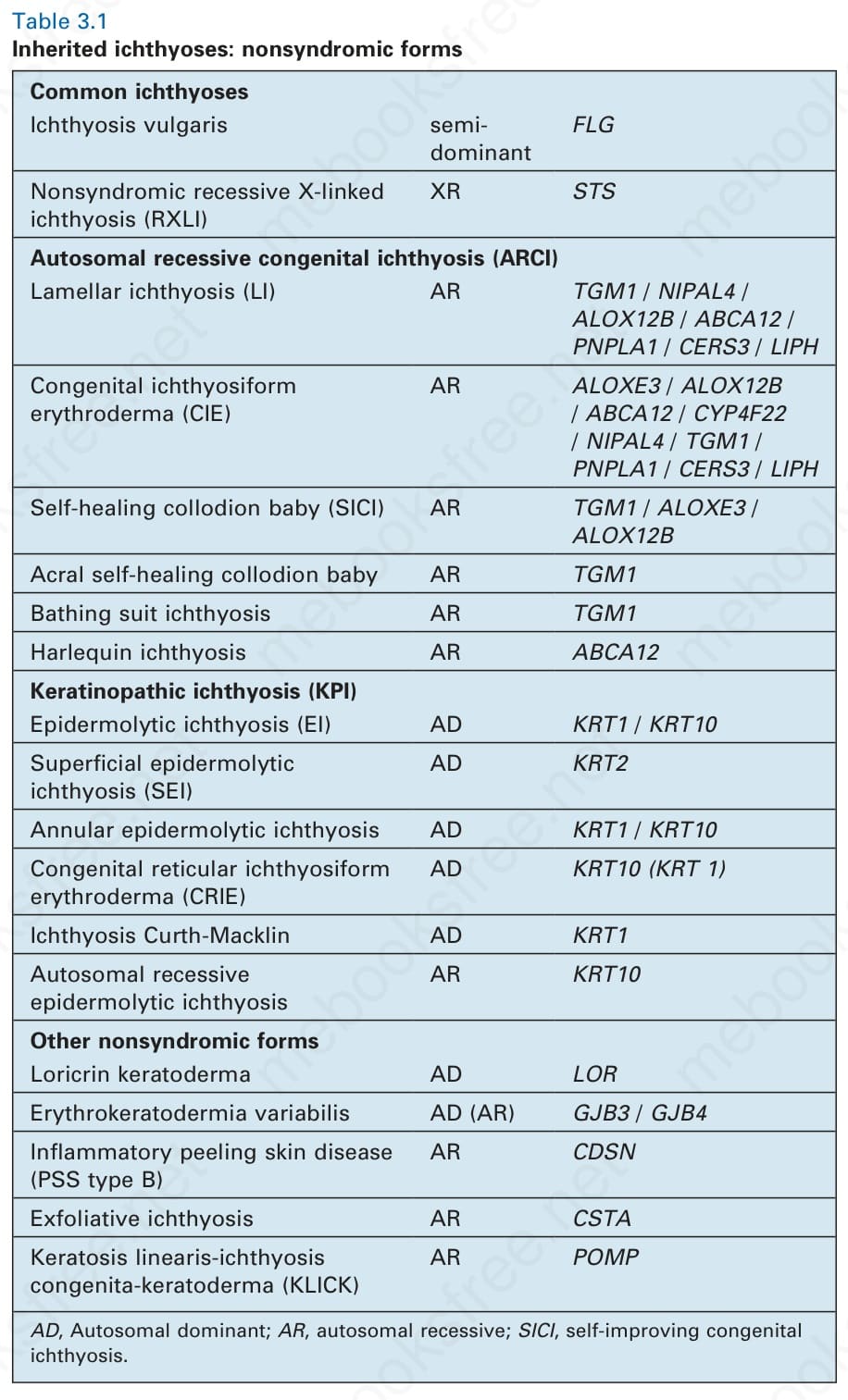
Table 3.1 Inherited ichthyoses: nonsyndromic forms
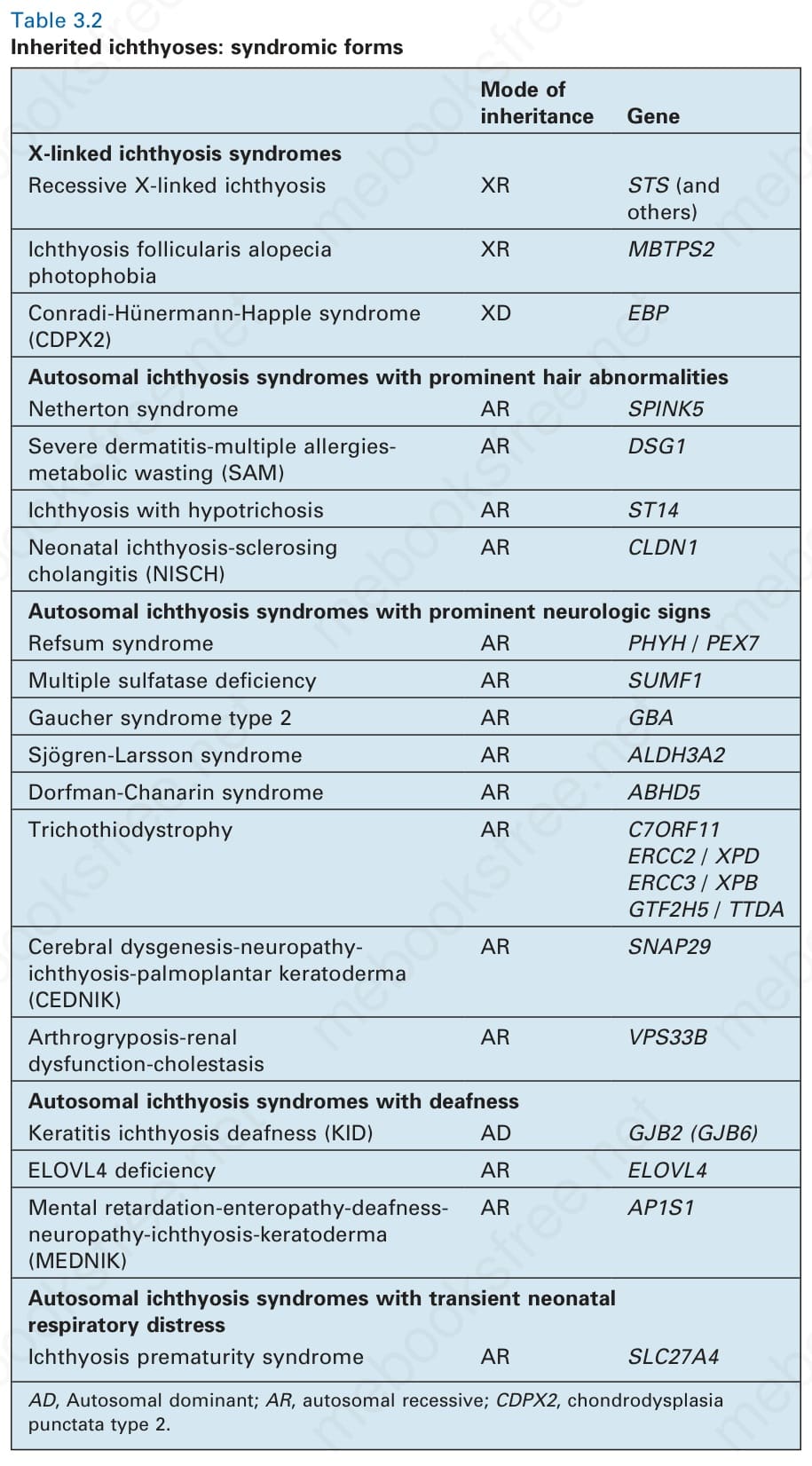
Table 3.2 Inherited ichthyoses: syndromic forms
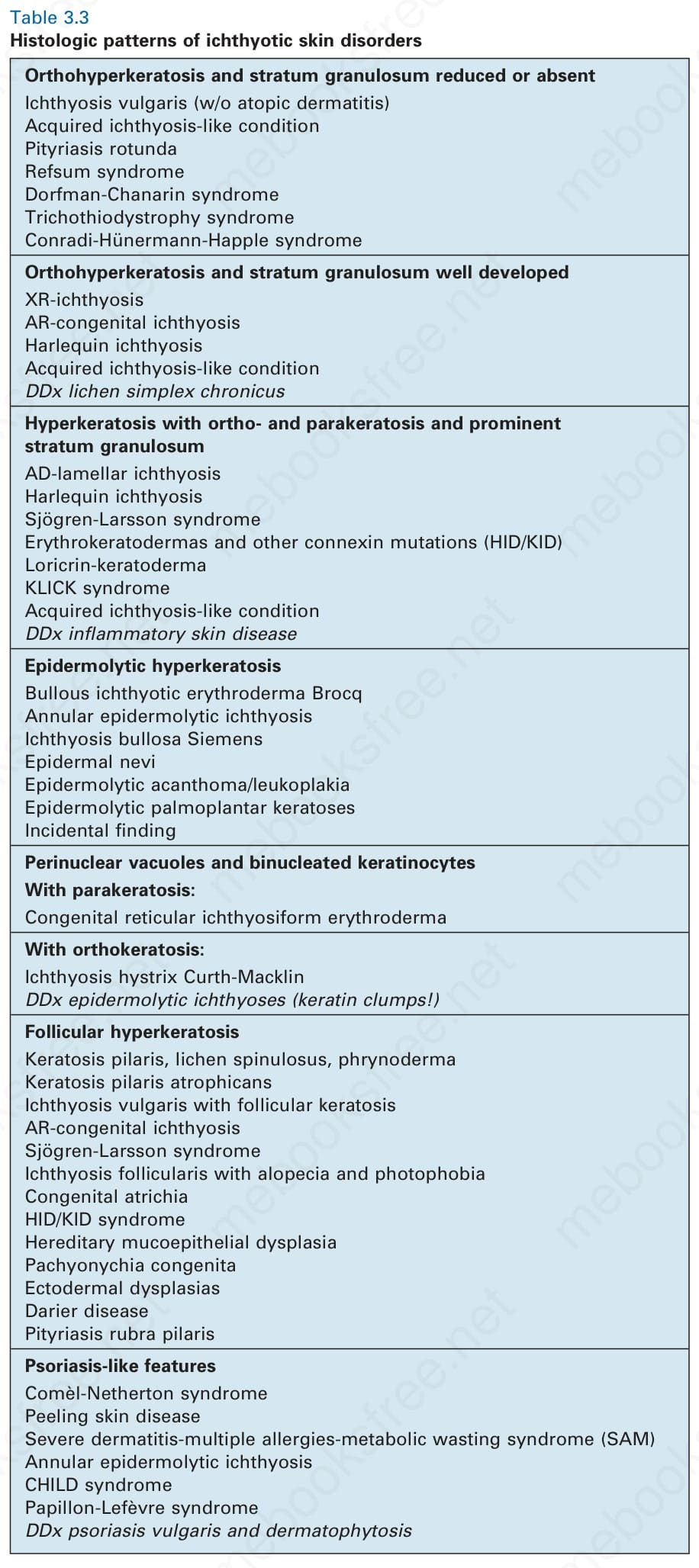
Table 3.3 Histologic patterns of ichthyotic skin disorders