Schwannoma–perineurioma 混合瘤 (Schwannoma–perineurioma hybrid)
Schwannoma–perineurioma 混合瘤 (schwannoma–perineurioma hybrid)
臨床特徵 (Clinical Features)
Schwannoma–perineurioma 混合瘤通常表現為一個無症狀的小型皮下、真皮內、或罕見更深部的結節,性別分布相當。
任何被發現有 neurofibroma 的病人,即使看似為孤立病灶,都必須仔細檢查是否有其他 neurofibromatosis 的徵象,這一點至關重要。Neurofibromas,特別是 neurofibromatosis type I 中的叢狀變異型 (plexiform variant),確實有一種無庸置疑、但並不常見的惡性轉變傾向,但這種轉變極為罕見。
1790 Connective tissue tumors
多核巨細胞 (multinucleated giant cells) 為 S100 protein 陰性、CD34 陽性,它們並不像先前所建議的那樣是 neurofibromatosis 診斷的線索。10–12 散布的發炎細胞、特別是肥大細胞 (mast cells),是一項顯著的特徵 (Fig. 35.328)。
基質膠原蛋白與黏液 (stromal collagen and mucin) 的相對含量在病灶內與病灶之間皆有所不同;膠原蛋白的玻璃樣變 (hyalinization) 有時可發生 (Fig. 35.329)。然而,看不到可辨識的雙相外觀 (biphasic appearance)(見 schwannoma)。某些病灶中存在顯著的硬化 (sclerosis)。
在較典型的皮膚/皮下普通型 neurofibromas 中。6 真正良性病灶的局部復發非常少見。
在近期一項針對 neurofibromatosis 1 病人的回溯性研究中,皮膚 neurofibromas 多數位於軀幹,其次為四肢及頭頸部。7 腫瘤數目被發現會隨年齡增加。
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
Neurofibroma 的顯微鏡特徵容易辨認。典型而言,它是一個相當界限清楚但無包膜 (unencapsulated) 的真皮或皮下病灶。與 schwannoma 不同,它含有眾多的小型神經纖維。
它由鬆散排列的梭形細胞 (spindled cells) 組成,胞質稀少而淡染、核呈細長波浪狀,置於纖維性、膠原性、有時為黏液樣的基質中 (Figs 35.325–35.327)。可出現多核花瓣狀巨細胞 (multinucleated floret-like giant cells),在少數腫瘤中數量眾多。8,9 這些
多核巨細胞為 S100 protein 陰性、CD34 陽性,它們並不像先前所建議的那樣是 neurofibromatosis 診斷的線索。10–12 散布的發炎細胞、特別是肥大細胞 (mast cells),是一項顯著的特徵 (Fig. 35.328)。
罕見可發現類似 glomus 小體的構造 (glomus-like bodies)。13 在瀰漫型 (diffuse) 與叢狀 (plexiform) 變異型中可見血管變化。14
退化性核多形性 (degenerative nuclear pleomorphism) 與深染 (hyperchromasia) 為罕見特徵(與 ancient schwannoma 相比),偶爾散發性 neurofibroma 會伴隨極低的有絲分裂活性(見下文)(Figs 35.330–35.332)。然而,在 neurofibromatosis type I 病人的病灶中出現此類多形性時,應促使對有絲分裂進行非常徹底的搜尋。在 neurofibromatosis type I 的情境下出現後者(有絲分裂),被視為惡性的證據。在近期一項共識中,「未定生物潛能之非典型神經纖維瘤性腫瘤 (atypical neurofibromatous neoplasms of uncertain biologic potential, ANNUBP)」一詞已被提出用於這些病灶(見下文 Variants 段落)。15
S100 protein 與 SOX10 的陽性染色僅見於 30–50% 的細胞中。16 也可見不等程度的 CD34、以及某些病例的 EMA 陽性。
1791 Benign neural tumors
在超微結構上,neurofibroma 由 Schwann 細胞、纖維母細胞 (fibroblasts) 與神經束膜細胞 (perineurial cells) 混合組成。17 Neurofibromas 中已證實有克隆性 (clonality),支持其為一種腫瘤性過程。17,18 雖然染色體不平衡 (chromosomal imbalances) 最常見於 neurofibromatosis type I 的 neurofibromas,但它們也已在散發性 neurofibromas 中被辨識出。19 染色體缺失是最常見的事件,特別是 NF1 基因所在的 17 號染色體與 19p。19,20 NF1 的功能喪失突變 (loss-of-function mutations) 也很常見,並見於多種 neurofibroma 細胞類型中。14 較近期的研究顯示,肥大細胞雖不帶有 NF1 突變,但在 neurofibromatosis 的小鼠模型中已被證明是 neurofibromas 的必要組成成分。15–26
近期已顯示 KIR2DL5 突變與缺失牽涉於散發性真皮 neurofibromas 的致病機轉中。27 已有數個基因特徵 (gene signatures) 被報告牽涉於 neurofibroma 及其惡性轉變的致病機轉。28–30 另一項研究將小型腫瘤血管的活化周細胞 (activated pericytes) 與平滑肌細胞牽涉於 neurofibromas 的致病機轉中。31
Variants
• Myxoid neurofibroma 是一種組織學變異型,不一定與 neurofibromatosis type I 相關,代表一種
1792 Connective tissue tumors
帶有廣泛基質黏液沉積的傳統型 neurofibroma。因此,病灶可能顯得明顯細胞稀少 (hypocellular) (Fig. 35.333)。1–3
• Plexiform neurofibroma 被視為 neurofibromatosis type I 的特異性病徵 (pathognomonic),最常表現於任一性別的兒童。1–3 其解剖分布不一,但最常見的部位是頭頸區。表淺覆蓋病灶的皮膚常顯示大而冗餘的皺褶,伴有不等程度的色素增生;其下方骨骼可能肥大。其大體外觀為一團呈複雜迂曲排列的神經纖維,令人聯想到「一袋蠕蟲 (bag of worms)」。組織學特徵由大而厚的神經或神經纖維組成,常在較典型 neurofibroma 的背景中顯示廣泛的黏液樣變化 (Figs 35.334 and 35.335)。然而,周圍組織有時可顯示瀰漫型 neurofibroma 的變化。顯示顯微鏡下叢狀型態的小型皮膚病灶不一定與 neurofibromatosis type I 相關。可辨識出類似脂肪母細胞、充滿黏液的細胞 (lipoblast-like mucin filled cells)。32,33 可見假腺體 (pseudoglandular) 或微囊狀 (microcystic) 成分。34
• Diffuse neurofibroma 在高達 20–30% 的病例中與 neurofibromatosis type I 相關,最常見於年輕病人,一般發生於頭、頸或軀幹。1–3 它表現為一個界限不清的皮下增厚區。組織學上,它的特徵為
帶有瀰漫浸潤性生長模式 (diffuse infiltrative growth pattern) 的神經纖維瘤性組織,其基質傾向於均勻膠原性而非黏液樣 (Figs 35.336 and 35.337)。Meissner 小體樣分化 (Meissnerian differentiation) 常是一項顯著的特徵 (Figs 35.338 and 35.339)。
• Pigmented neurofibroma 的特徵為類似 Meissner 小體的旋渦狀構造 (whorled structures),伴有散布、對黑色素細胞標記 (melanocytic markers) 呈陽性的色素細胞 (Fig. 35.340)。35–37 在一位病人中,曾記錄到與多毛症 (hypertrichosis) 的關聯。38
• Granular cell neurofibroma 局部由具有豐富 periodic acid-Schiff 陽性、抗 diastase 顆粒狀胞質的腫瘤細胞組成。3 診斷常依賴於辨識出具有較典型型態的區域。
• Epithelioid neurofibroma 局部由具有粉紅色胞質的上皮樣細胞 (epithelioid cells) 組成,背景則為其餘呈典型的 neurofibroma。
• Dendritic cell neurofibroma with pseudorosettes 是 neurofibroma 一種獨特的變異型,具有結節狀生長模式與兩種細胞類型。39 小而圓、深染、類似淋巴球的深色細胞圍繞著較大的細胞,後者具有泡狀核 (vesicular nuclei)、頻繁的核內包涵體 (intranuclear inclusions) 與豐富的淡染胞質,形成獨特的假玫瑰花結外觀 (pseudorosette appearance) (Figs 35.341 and 35.342)。兩種細胞類型皆對 S100 protein 與 CD57 呈陽性。40 已有一例具肉芽腫性 (granulomatous) 模式的病例被報告。41 An
1793 Benign neural tumors
1794 Connective tissue tumors
近期已記錄到此變異型的一例神經內 (intraneural) 病例。42 已描述過一例口腔內 (intraoral) 病例。43 類似病灶可見於 neurofibromatosis type I。44
• Cellular neurofibroma with atypia (atypical neurofibroma) 指一種散發性 neurofibroma,具有增加的細胞密度、局灶性非典型 (focal atypia) 與極低的有絲分裂活性。45,46 這些病灶似乎呈良性行為。然而,類似病灶若處於 neurofibromatosis type I 的情境且存在任何有絲分裂活性,則應被視為惡性的證據。
• Pacinian neurofibroma 最好被視為 schwannoma 的一種變異型(見上文)。過去被描述為 pacinian neurofibromas 的範例其實是神經鞘黏液瘤 (nerve sheath myxomas)。
• Lipomatous neurofibroma 指 neurofibroma 內出現成熟脂肪細胞 (mature fat cells) 的聚集。47–49 它似乎在頭頸部較常見。50 與伴有脂肪化生 (fatty metaplasia) 的神經化痣 (neurotized nevus) 的區別可能無法做到(見鑑別診斷)。
• Hybrid tumors(混合瘤)同時顯示 neurofibroma 與 schwannoma 的特徵;罕見可見 perineurioma 的特徵。51–53 也已記錄到神經束膜分化 (perineurial differentiation) 與神經內發生 (intraneural occurrence)。54,55 在一部分混合瘤中已辨識出單體 22 (Monosomy 22)。56 此外,已顯示混合瘤與 neurofibromatosis 及 schwannomatosis 有強烈關聯。57
• 已報告過單一一例具有透明細胞變化 (clear cell change)、一例具有氣球細胞變化 (balloon cell change)、以及一例具有顯著硬化而類似硬化性纖維瘤 (sclerotic fibroma) 的病例。58–61 已記錄過一種被描述為 angioneurofibroma 的變異型。62
• Atypical neurofibromatous neoplasms of uncertain biologic potential (ANNUBP)(未定生物潛能之非典型神經纖維瘤性腫瘤)是一個被提出的術語,用於先前被不一致地描述為 atypical neurofibromas 或低度惡性周邊神經鞘瘤 (low-grade malignant peripheral nerve sheath tumors) 的 neurofibromas。14,63 根據該共識報告,單憑核非典型 (nuclear atypia) 不足以診斷惡性。然而,具有細胞學非典型 (cytologic atypia) 並伴隨喪失通常 neurofibroma 結構、高細胞密度、且/或有絲分裂活性高於每 50 HPF 1 個但低於每 10 HPF 3 個的腫瘤令人擔憂。因此有人提議,當腫瘤顯示上述標準中的至少兩項時,應使用「未定生物潛能之神經纖維瘤性腫瘤 (neurofibromatous neoplasms of uncertain biologic potential)」此一名稱。其中部分腫瘤可能顯示 S100 protein 與 SOX10 免疫反應性減弱、p16/CDKN2A 表現喪失、Ki67 升高、以及如惡性周邊神經鞘瘤所見的 TP53 過度表現。此外,如後者半數中所描述的,也可見到三甲基化組蛋白 3 離胺酸 27 (trimethylated histone 3 lysine 27) 表現的完全喪失。14
• 中樞型或聽神經型、或第二型 neurofibromatosis (NF2;NF2 基因編碼位於 22q12.2 的 merlin 蛋白)。也已描述過第三種變異型——節段性 neurofibromatosis (segmental neurofibromatosis)。它是由於 NF1 或 NF2(較常為前者)的鑲嵌現象 (mosaicism) 所造成。
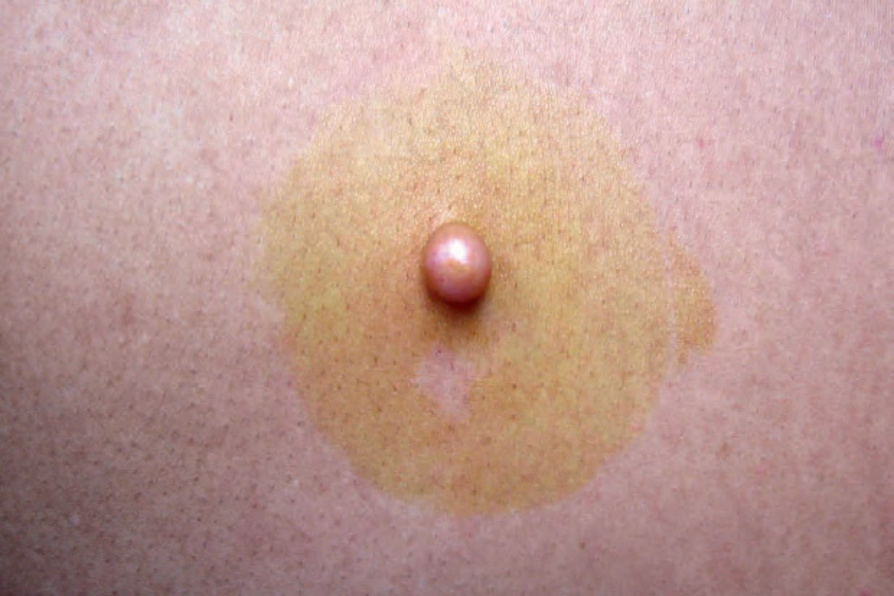
圖 35-324:Neurofibroma:此例表現為一個界限清楚、堅實的外生性結節 (circumscribed firm exophytic nodule)。By courtesy of J. Dayrit, MD, Manila, The Philippines.
Fig. 35.324 Neurofibroma: this example presented as a circumscribed firm exophytic nodule. By courtesy of J. Dayrit, MD, Manila, The Philippines.
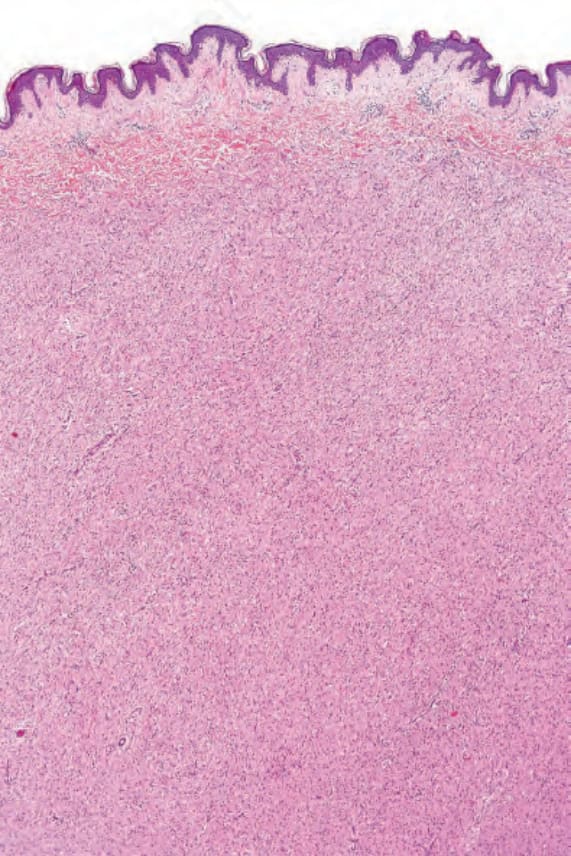
圖 35-325:Neurofibroma:腫瘤由細胞邊界不清的小型梭形細胞 (small spindled cells) 組成。
Fig. 35.325 Neurofibroma: the tumor is composed of small spindled cells with indistinct cell borders.
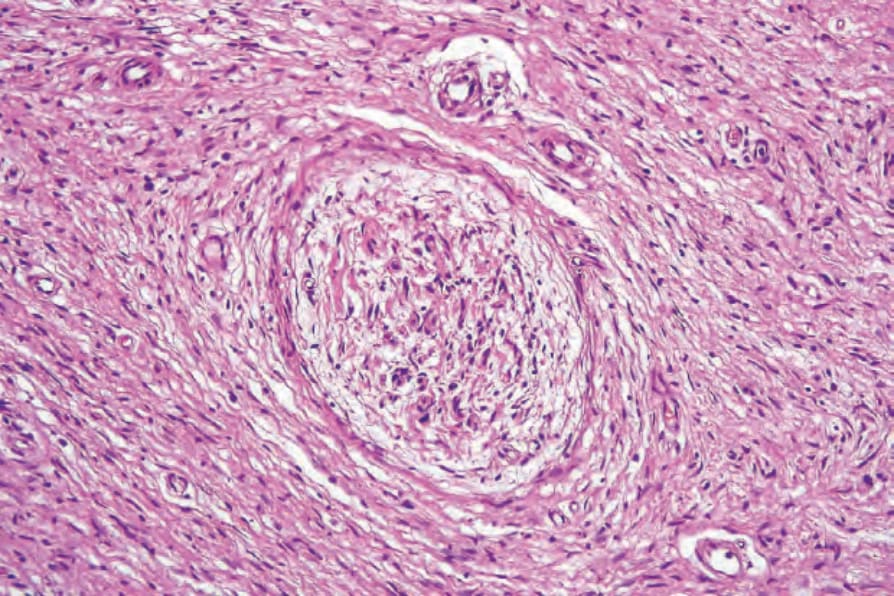
圖 35-326:Neurofibroma:視野中央可見一條大型神經幹 (large nerve trunk)。
Fig. 35.326 Neurofibroma: a large nerve trunk is present in the center of the field.
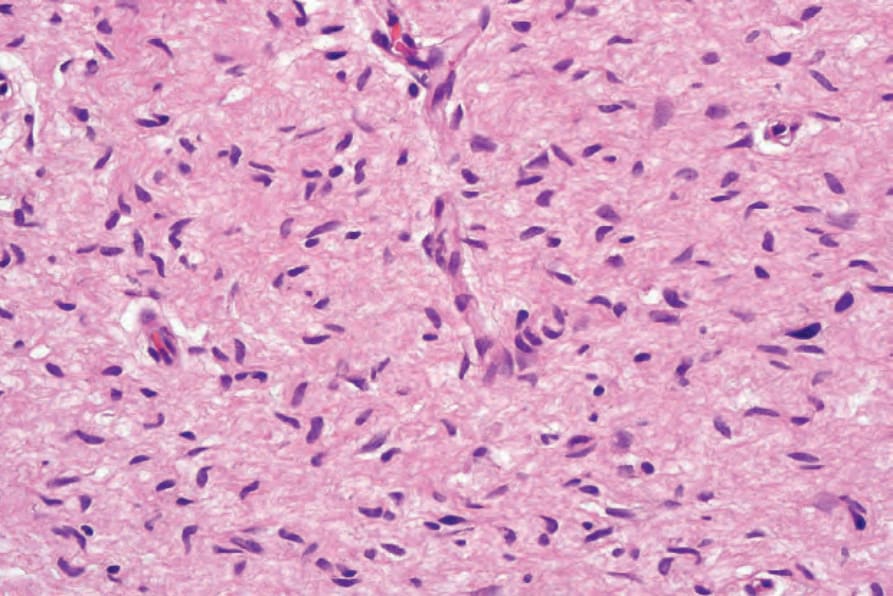
圖 35-327:Neurofibroma:梭形細胞的細胞核具有特徵性的細長波浪狀 (elongated and wavy)。
Fig. 35.327 Neurofibroma: the nuclei of the spindled cells are characteristically elongated and wavy.
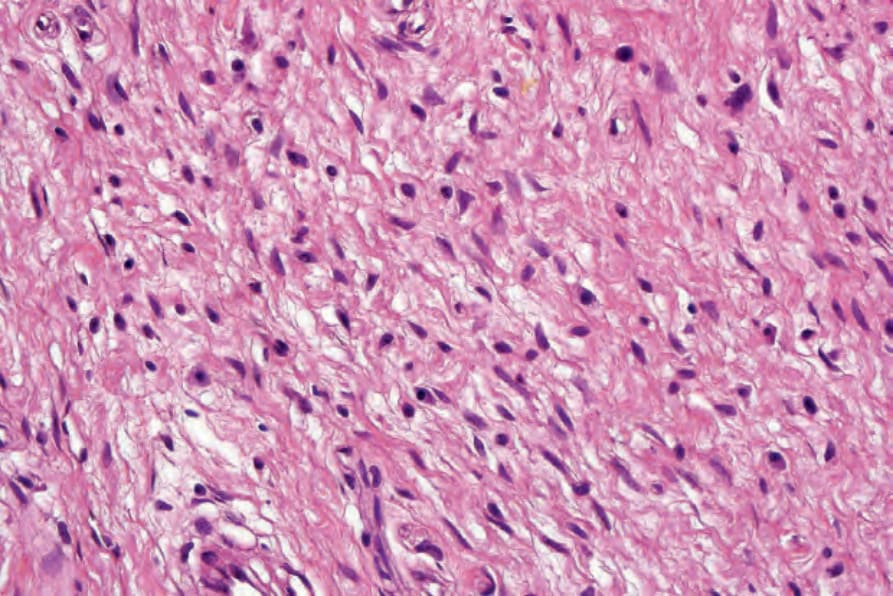
圖 35-328:Neurofibroma:這些腫瘤中常見具有顆粒狀嗜伊紅胞質的肥大細胞 (mast cells)。
Fig. 35.328 Neurofibroma: mast cells with granular eosinophilic cytoplasm are frequently seen in these tumors.
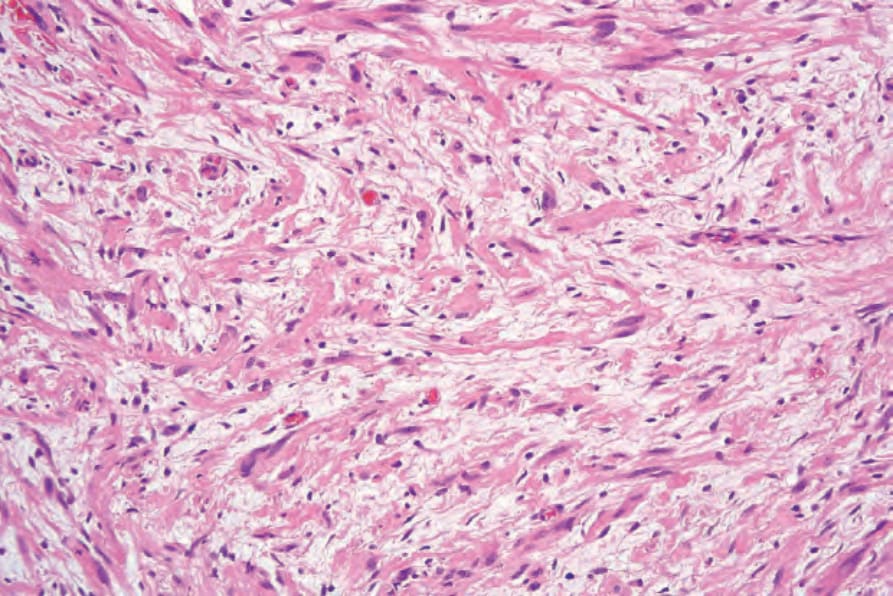
圖 35-329:Neurofibroma:膠原蛋白含量變異極大,但可能很顯著,如本例所示。
Fig. 35.329 Neurofibroma: the collagen content is highly variable, but may be prominent, as in this example.
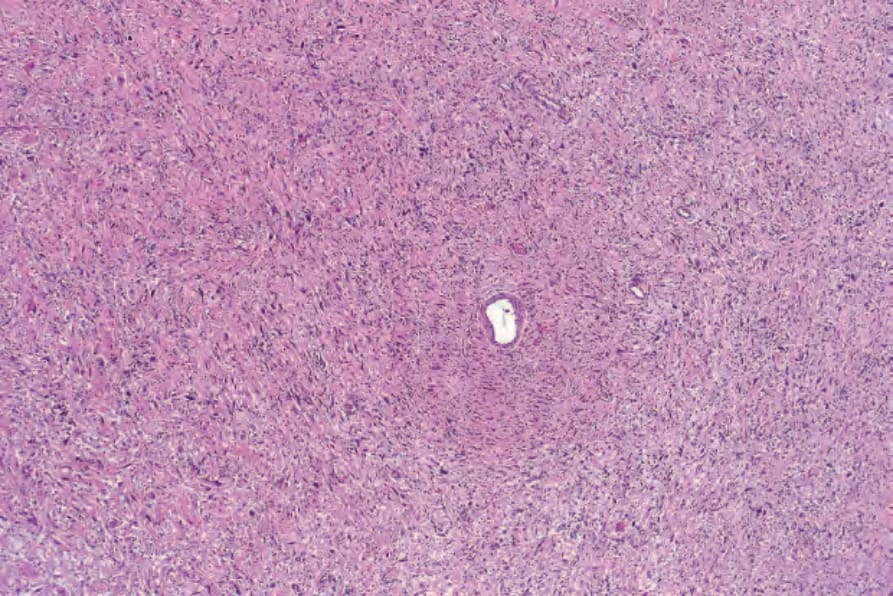
圖 35-330:Atypical neurofibroma:即使在此放大倍率下,深染且增大的細胞核 (hyperchromatic and enlarged nuclei) 也清晰可見。
Fig. 35.330 Atypical neurofibroma: even at this magnification, hyperchromatic and enlarged nuclei are evident.
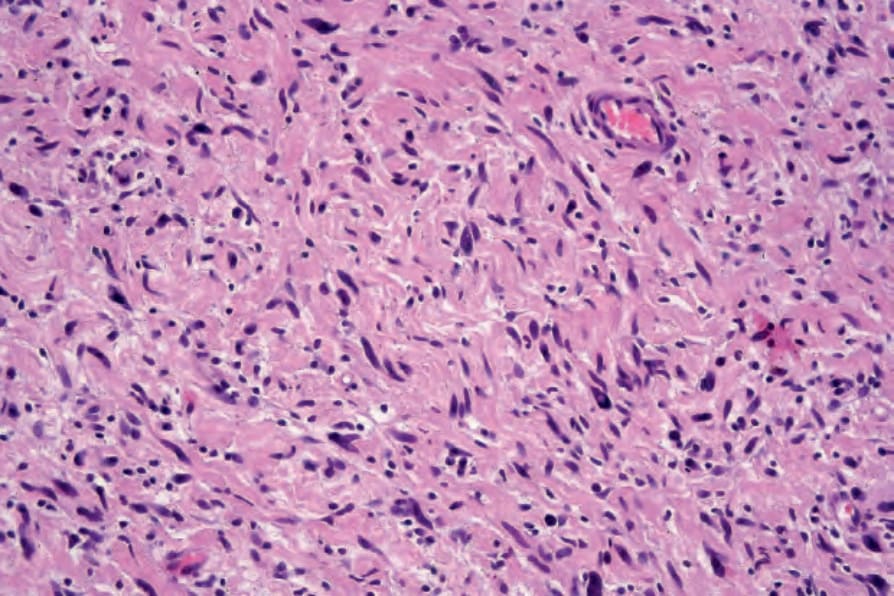
圖 35-331:Atypical neurofibroma:高倍視野顯示多形性核 (pleomorphic nuclei)。
Fig. 35.331 Atypical neurofibroma: high-power view showing pleomorphic nuclei.
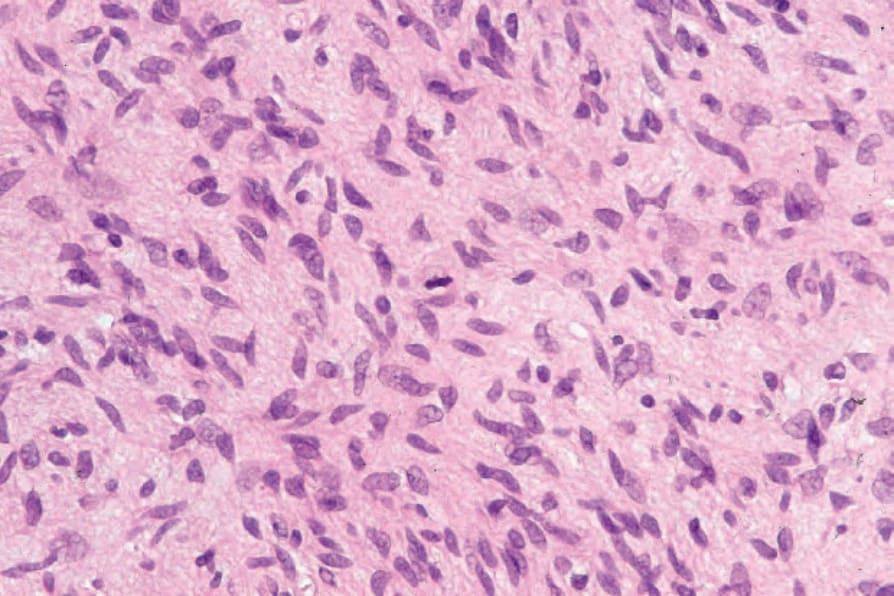
圖 35-332:Atypical neurofibroma:極偶爾地,可辨識出單個有絲分裂 (single mitoses)。
Fig. 35.332 Atypical neurofibroma: very occasionally, single mitoses may be identified.
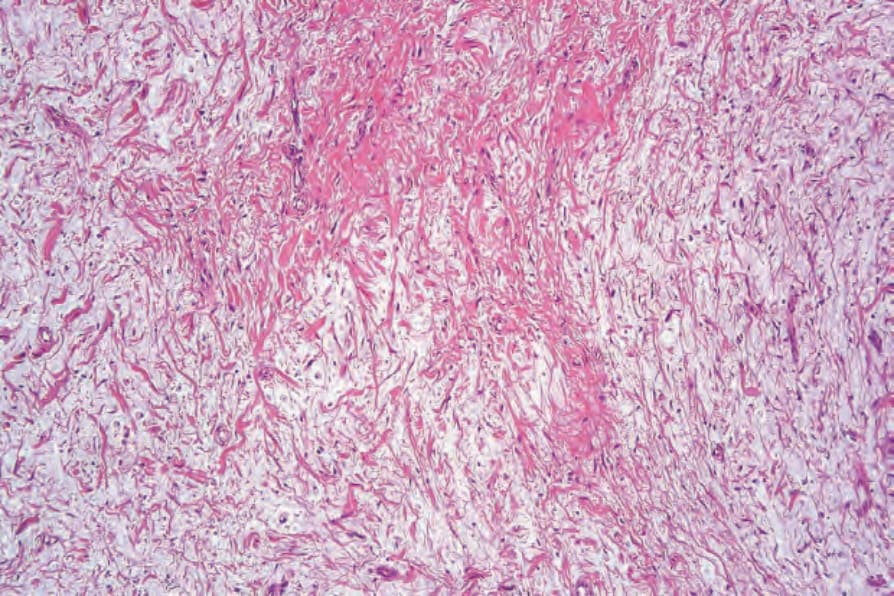
圖 35-333:Myxoid neurofibroma:在此變異型中,存在顯著的基質黏液 (stromal mucin)。
Fig. 35.333 Myxoid neurofibroma: in this variant, there is marked stromal mucin.
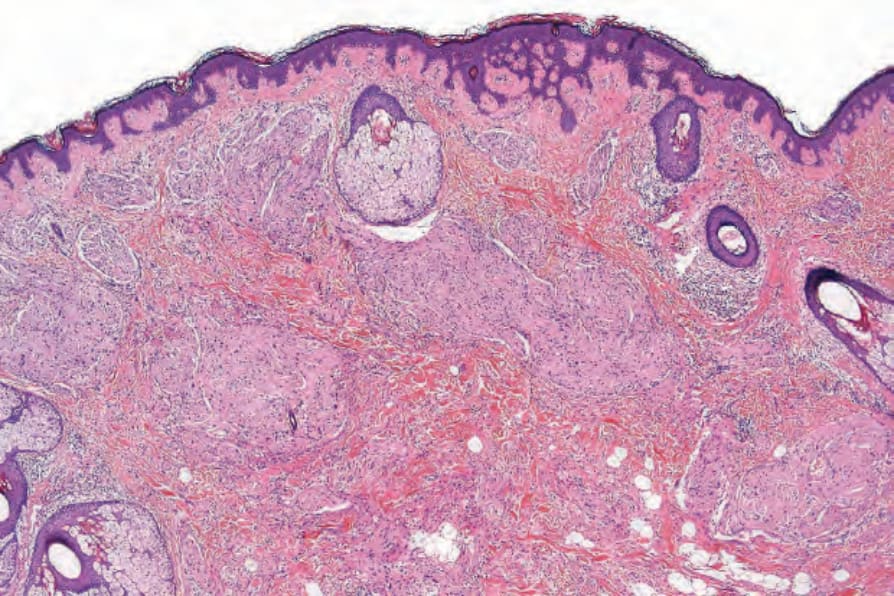
圖 35-334:Plexiform neurofibroma:增厚、雜亂分布的神經幹 (nerve trunks) 存在於網狀真皮 (reticular dermis) 中。
Fig. 35.334 Plexiform neurofibroma: thickened, haphazardly distributed nerve trunks are present in the reticular dermis.
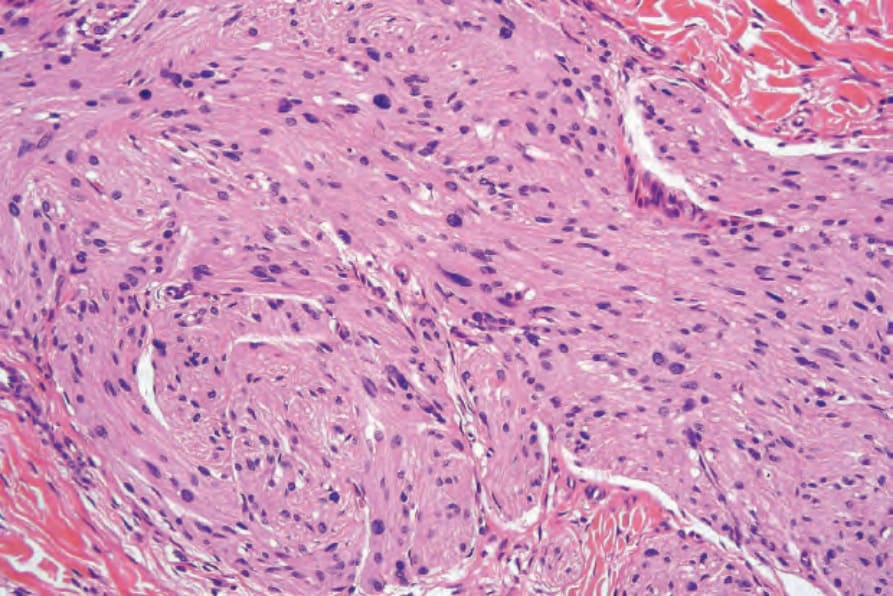
圖 35-335:Plexiform neurofibroma:可見肥大的神經 (hypertrophied nerves) 嵌埋於纖維母細胞與 Schwann 細胞的基質中。
Fig. 35.335 Plexiform neurofibroma: hypertrophied nerves are seen embedded in a matrix of fibroblasts and Schwann cells.
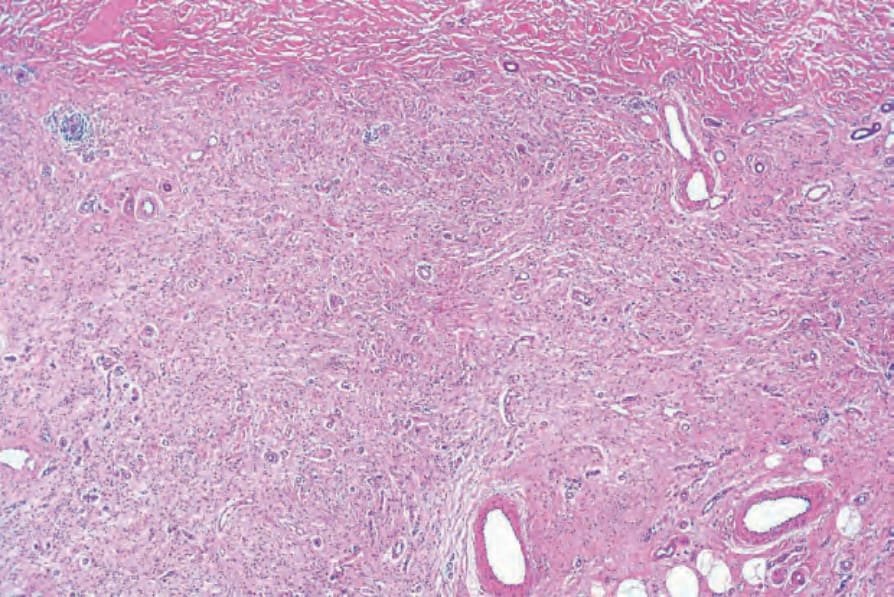
圖 35-336:Diffuse neurofibroma:乳頭層與網狀真皮皆被神經纖維瘤性組織廣泛浸潤。
Fig. 35.336 Diffuse neurofibroma: both the papillary and the reticular dermis are extensively infiltrated by neurofibromatous tissue.
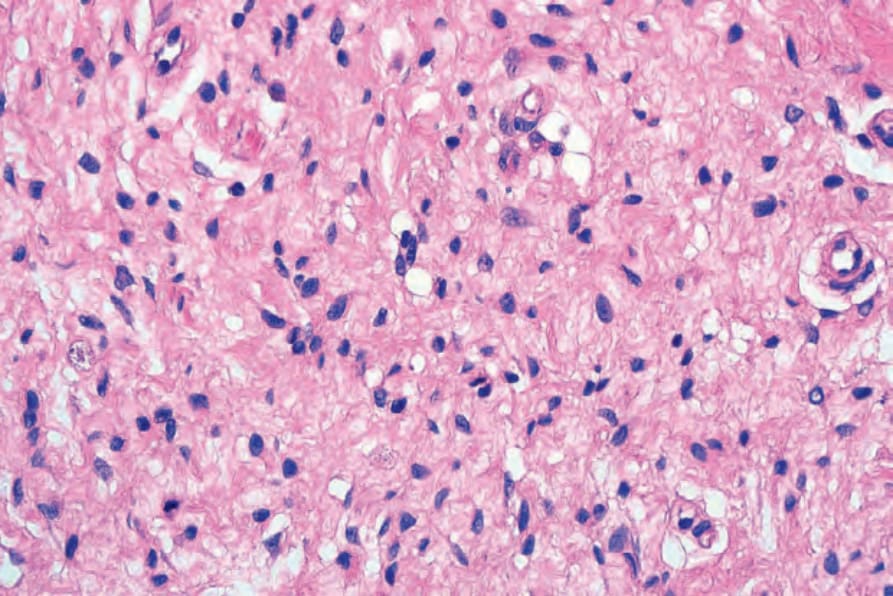
圖 35-337:Diffuse neurofibroma:高倍視野。
Fig. 35.337 Diffuse neurofibroma: high-power view.
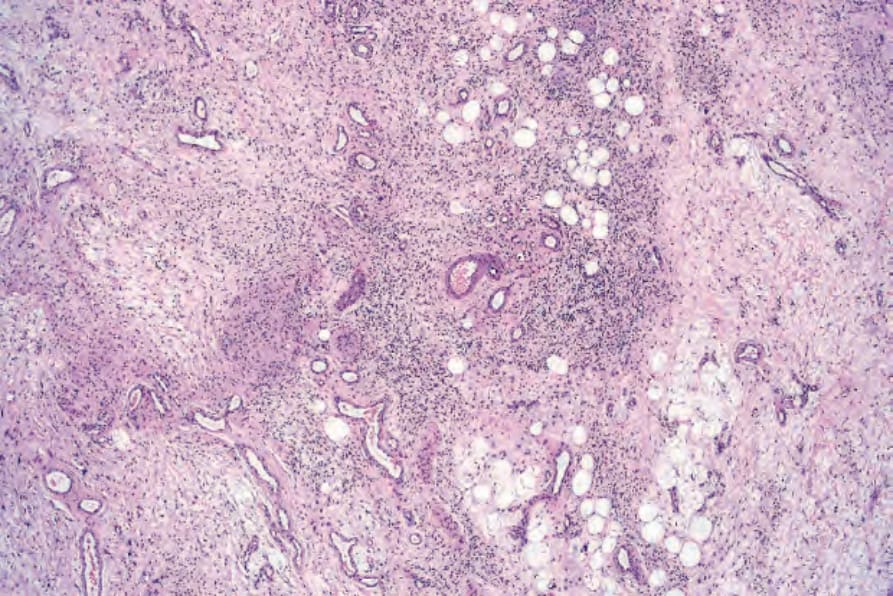
圖 35-338:Diffuse neurofibroma:存在顯著的類器官分化 (organoid differentiation)。
Fig. 35.338 Diffuse neurofibroma: there is marked organoid differentiation.
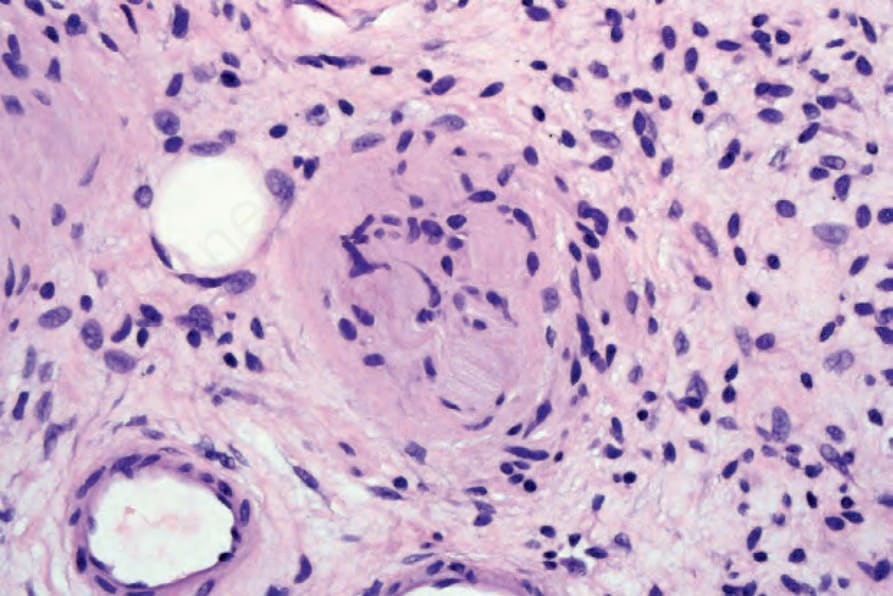
圖 35-339:Diffuse neurofibroma:視野中央可見朝向 Meissner 小體 (Meissner’s corpuscle) 的分化。
Fig. 35.339 Diffuse neurofibroma: in the center of the field there is differentiation towards a Meissner’s corpuscle.
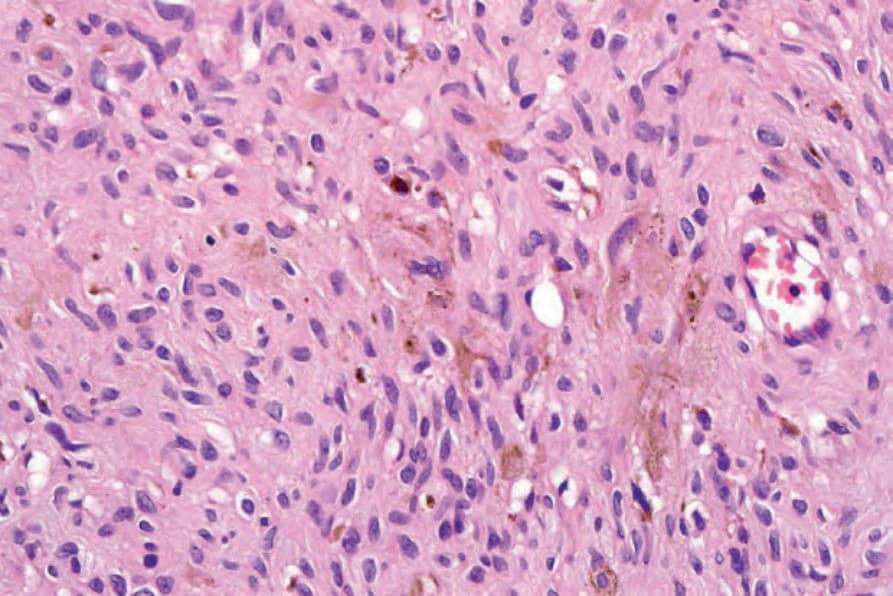
圖 35-340:Pigmented neurofibroma:注意色素性樹突狀細胞 (pigmented dendritic cells)。By courtesy of H. Diwan, MD, Houston, Texas, USA.
Fig. 35.340 Pigmented neurofibroma: note the pigmented dendritic cells. By courtesy of H. Diwan, MD, Houston, Texas, USA.
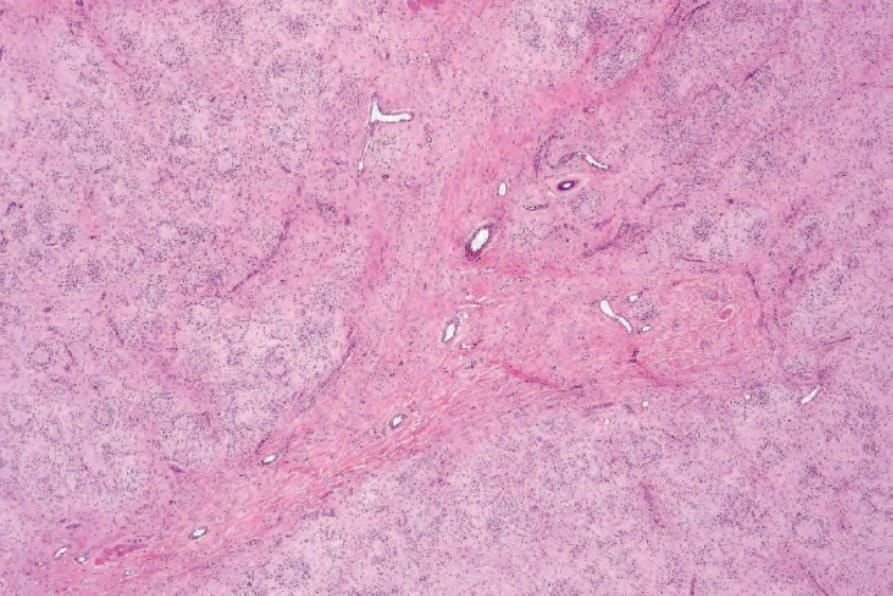
圖 35-341:Neurofibroma with pseudorosettes:低倍視野顯示明顯的假玫瑰花結 (pseudorosettes)。
Fig. 35.341 Neurofibroma with pseudorosettes: low-power view showing conspicuous pseudorosettes.
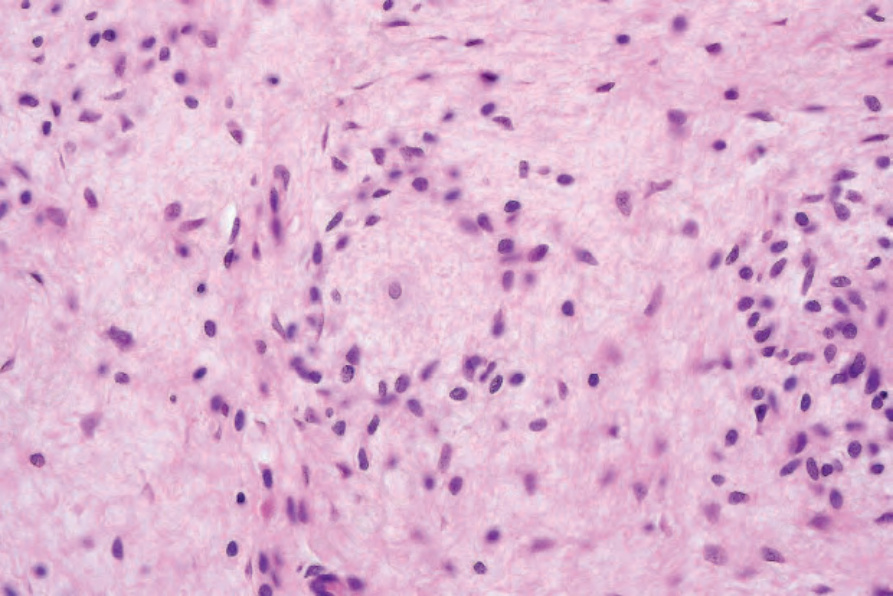
圖 35-342:Neurofibroma with pseudorosettes:有一個位於中央的大型淡染細胞,周圍環繞著一圈小型類淋巴球細胞 (lymphocyte-like cells)。
Fig. 35.342 Neurofibroma with pseudorosettes: there is a single central large pale cell surrounded by a mantle of small lymphocyte-like cells.
Neurofibromatosis 的其他變異型也已被記錄,包括遺傳性脊髓 neurofibromatosis (hereditary spinal neurofibromatosis)、schwannomatosis、家族性腸道 neurofibromatosis (familial intestinal neurofibromatosis)、體染色體顯性「單純咖啡牛奶斑 (café-au-lait spots alone)」、體染色體顯性「單純 neurofibromas (neurofibromas alone)」、Watson 症候群、Noonan/neurofibromatosis 症候群、以及多發性痣、多發性 schwannomas、與多發性陰道平滑肌瘤 (multiple vaginal leiomyomas)。1,2
Neurofibromatosis type I 是一種重要的先天性神經皮膚疾病 (neurocutaneous disorder),呈體染色體顯性遺傳模式,影響約 1/3000 活產嬰兒;亦有若干病例因自發性生殖細胞突變 (spontaneous germline mutation) 而發生。3–12 它涵蓋一系列徵象與症狀,並可能侵犯身體大多數系統。NF1 的基因已被選殖定位於 17q11.2 染色體。13–16 所編碼的蛋白稱為 neurofibromin,協助控制細胞內的 Ras 活性。17–19 腫瘤在 NF1 野生型對偶基因 (wild type allele) 發生異型合子性喪失 (loss of heterozygosity) 時產生。染色體不平衡在 NF1 相關 neurofibromas 中比在散發性 neurofibromas 中更常見。20 在 NF1 中所描述的其他染色體不平衡包括 19 號與 22q 染色體的缺失。20 另見上文 neurofibroma 段落。
具 NF1 但無皮膚 neurofibromas 證據的病人,於 NF1 基因 exon 17 中帶有一個 3-bp 框內缺失 (inframe deletion)。21
近期已描述一種類 neurofibromatosis type 1 的體染色體顯性症候群,其不帶有 NF1 突變,但與 SPRED1 (sprout-related EVH1 domain-containing protein 1) 的生殖細胞失活突變 (germline inactivating mutations) 相關。22 在此症候群中,病人表現為咖啡牛奶斑 (café-au-lait macules)、腋窩雀斑 (axillary freckling) 與巨頭症 (macrocephaly)。
美國國家衛生研究院 (National Institutes of Health, NIH) 對 NF1 的診斷標準包括以下兩項或以上:23
• 六個或以上的咖啡牛奶斑,在 6 歲以下兒童直徑大於 5.0 mm、在較年長個體大於 15 mm,
• 任何類型的兩個或以上 neurofibromas、或一個 plexiform neurofibroma,
• 腋窩或腹股溝區的雀斑,
• 一個視神經膠質瘤 (optic nerve glioma),
• 兩個或以上的 Lisch 結節 (Lisch nodules)(虹膜錯構瘤,iris hamartomas),
• 一處獨特的骨性病灶,如蝶骨發育不良 (dysplasia of the sphenoid bone) 或長骨皮質變薄,伴或不伴假關節 (pseudoarthrosis),
• 一位罹患 NF1 的一等親。Neurofibromatosis type I 顯示非常廣泛的臨床變異性。一項大型研究已證實受影響的先證者 (probands) 中數對特徵之間的關聯:
• 間擦部位雀斑 (intertriginous freckling) 與 Lisch 結節,
• 散在性 neurofibromas 與 plexiform neurofibromas,
• 散在性 neurofibromas 與 Lisch 結節,
• plexiform neurofibromas 與脊柱側彎 (scoliosis),
• 學習障礙或智能不足與癲癇發作 (seizures)。24
鑑別診斷 (Differential Diagnosis)
斑塊期隆突性皮膚纖維肉瘤 (plaque-stage dermatofibrosarcoma protuberans) 的小型切片可能難以與 neurofibroma 區別。然而前者具有獨特的蕾絲狀脂肪浸潤模式 (lace-like pattern of infiltration of the fat),其真皮束傾向於與表皮平行,且腫瘤細胞對 S100 protein 陰性、CD34 陽性。若無殘餘痣細胞或表皮成分存在,舊的神經化痣 (neurotized nevi) 常與 neurofibroma 無法區別。兩種病灶中 S100 protein 皆呈陽性,但神經化痣傾向於對稱,且也較常為 NSE 陽性。重要的是,在罹患 neurofibromatosis 的病人中發生的 neurofibromas(包括上皮樣變異型)可見黑色素細胞標記的表現。64
咖啡牛奶斑 (café-au-lait macules) 為扁平、淡棕色的病灶,可分布於體表任何處,但主要見於身體非曝光面 (Fig. 35.343)。高達 10% 的人口可能自出生即有孤立性病灶。病人也可能表現出覆蓋於皮膚 plexiform neurofibromas 之上、顏色較深的斑 (Fig. 35.344)。有人提出真皮纖維母細胞衍生的幹細胞因子 (stem cell factor) 與肝細胞生長因子 (hepatocyte growth factor) 可能在此色素增生的發生中扮演角色。25
已有報告使用 CD34 作為區別 neurofibromas(neurofibroma 所見的特徵性指紋狀模式 (fingerprint pattern),相對於 melanoma)與梭形細胞黑色素瘤 (spindle cell melanomas) 的工具,但它並非完全可靠的特徵。65–67
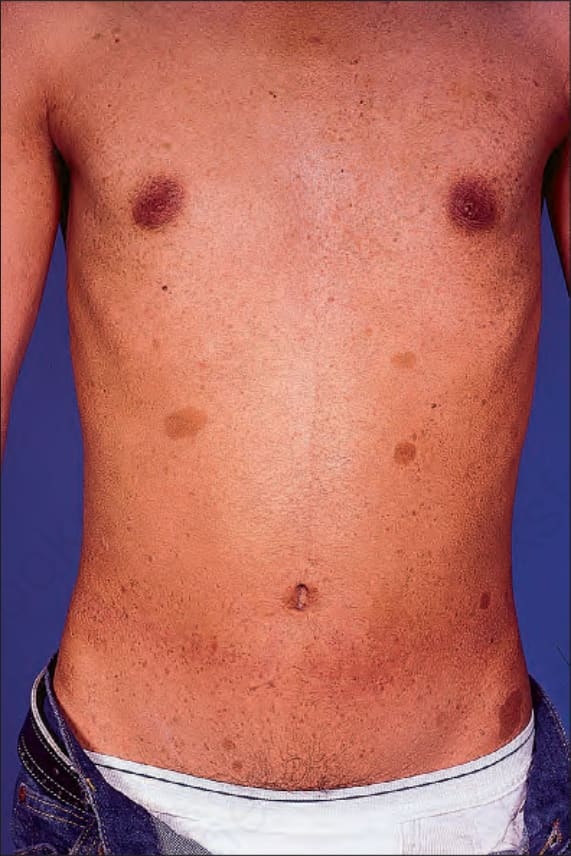
圖 35-343:Type I neurofibromatosis:典型咖啡牛奶斑 (café-au-lait macules) 的出現具有特徵性。By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Fig. 35.343 Type I neurofibromatosis: the presence of typical café-au-lait macules is characteristic. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
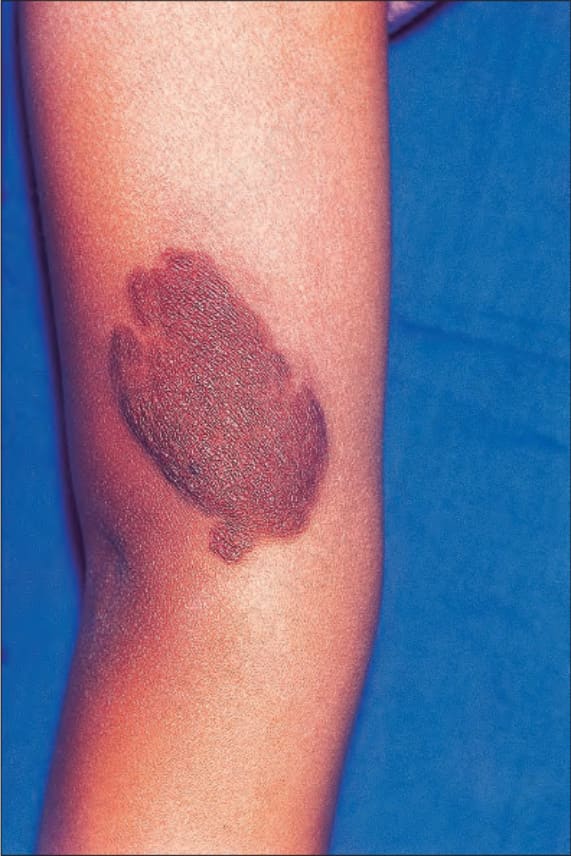
圖 35-344:Type I neurofibromatosis:此重度色素性隆起病灶覆蓋於一個皮膚 plexiform neurofibroma 之上。By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Fig. 35.344 Type I neurofibromatosis: this heavily pigmented raised lesion overlies a cutaneous plexiform neurofibroma. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.