Schwannoma–perineurioma hybrid
Schwannoma–perineurioma hybrid
Clinical features Hybrid Schwannoma–perineurioma usually presents as an asymptomatic small subcutaneous, dermal or rarely deeper nodule, with an equal sex
It is essential that any patient found to have a neurofibroma, even if seemingly in isolation, should be carefully examined for other stigmata of neurofibromatosis. Neurofibromas, particularly the plexiform variant in neurofibromatosis type I, have an undoubted, albeit uncommon, tendency to undergo malignant change, but such transformation is exceedingly rare
1790 Connective tissue tumors
multinucleated giant cells are S100 protein negative and CD34 positive and they are not a clue to the diagnosis of neurofibromatosis, as has previously been suggested.10–12 Scattered inflammatory cells, particularly mast cells, are a prominent feature (Fig. 35.328).
The relative amounts of stromal collagen and mucin vary both within and between lesions; hyalinization of collagen may sometimes occur (Fig. 35.329). However, no recognizable biphasic appearance is seen (see schwannoma). Prominent sclerosis is present in some lesions.
in the more typical cutaneous/subcutaneous ordinary neurofibromas.6 Local recurrence of truly benign lesions is very infrequent.
In a recent retrospective study in patients with neurofibromatosis 1, cutaneous neurofibromas were mostly located on the trunk followed by limbs and head and neck.7 The number of tumors was found to increase with age.
Pathogenesis and histologic features The microscopic features of neurofibroma are readily recognizable. Typically, it is a reasonably well-defined but unencapsulated dermal or subcutaneous lesion. In contrast to a schwannoma, it contains numerous small nerve fibers.
It consists of loosely arranged spindled cells with scanty pale cytoplasm and elongated wavy nuclei set in a fibrillar, collagenous and sometimes myxoid stroma (Figs 35.325–35.327). Multinucleated floret-like giant cells can be present, and in a small number of tumors are numerous.8,9 These
Glomus-like bodies are rarely found.13 Vascular changes may be seen in the diffuse and plexiform variants.14
Degenerative nuclear pleomorphism and hyperchromasia are rare features (compare with ancient schwannoma) and occasionally sporadic neurofibroma is associated with very low mitotic activity (see below) (Figs 35.330–35.332). Such pleomorphism in a lesion from a patient with neurofibromatosis type I, however, should prompt a very thorough search for mitoses. The presence of the latter in the setting of neurofibromatosis type I is regarded as evidence of malignancy. In a recent consensus the term ‘atypical neurofibromatous neoplasms of uncertain biologic potential (ANNUBP)’ has been proposed for these lesions (see below under Variants).15
Positive staining for S100 protein and SOX10 is seen in only 30–50% of cells.16 Variable CD34 and in some cases EMA positivity is also seen.
1791 Benign neural tumors
Ultrastructurally, a neurofibroma is composed of an admixture of Schwann cells, fibroblasts and perineurial cells.17 Clonality has been demonstrated in neurofibromas, favoring a neoplastic process.17,18 Although chromosomal imbalances are most frequently found in neurofibromas, in neurofibromatosis type I they have also been identified in sporadic neurofibromas.19 Loss of chromosomes is the most frequent event, particularly chromosomes 17 where the NF1 gene is located and 19p.19,20 Loss-of-function mutations in NF1 are also common and seen in multiple neurofibroma cell types.14 More recently, mast cells, while not having NF1 mutations, have been shown to be a required component of neurofibromas in mouse models of neurofibromatosis.15–26
Recently it has been shown that KIR2DL5 mutation and loss is implicated in the pathogenesis of sporadic dermal neurofibromas.27 Several gene signatures have been reported to be implicated in pathogenesis of neurofibroma and its malignant transformation.28–30 Another study has implicated activated pericytes and smooth muscle cells of small tumor vessels in the pathogenesis of neurofibromas.31
Variants
• Myxoid neurofibroma is a histologic variant not necessarily associated with neurofibromatosis type I, and represents a conventional
1792 Connective tissue tumors
neurofibroma with extensive deposition of stromal mucin. As a consequence, the lesion may appear markedly hypocellular (Fig. 35.333).1–3
• Plexiform neurofibroma, which is considered pathognomonic of neurofibromatosis type I, most often presents in children of either sex.1–-3 Its anatomical distribution varies, but the most common site is the head and neck area. Commonly, the skin overlying the lesion shows large and redundant folds with variable hyperpigmentation; the underlying bone may be hypertrophic. The macroscopic appearance is that of a mass of nerve fibers in complex and tortuous arrangement reminiscent of a bag of worms. The histologic features consist of large thick nerves or nerve fibers often showing extensive myxoid change within a background of more typical neurofibroma (Figs 35.334 and 35.335). The surrounding tissue, however, may sometimes show changes of a diffuse neurofibroma. Small cutaneous lesions showing a microscopic plexiform pattern are not necessarily associated with neurofibromatosis type I. Lipoblast-like mucin filled cells may be identified.32,33 Pseudoglandular or microcystic elements may be seen.34
• Diffuse neurofibroma, which is associated with neurofibromatosis type I in up to 20–30% of cases, is most often seen in young patients and generally occurs on the head, neck or trunk.1–3 It presents as an ill-defined area of subcutaneous thickening. Histologically, it is
characterized by neurofibromatous tissue with a diffuse infiltrative growth pattern in which the stroma tends to be uniformly collagenous rather than myxoid (Figs 35.336 and 35.337). Meissnerian differentiation is often a prominent feature (Figs 35.338 and 35.339).
• Pigmented neurofibroma is characterized by whorled structures similar to Meissner corpuscles with scattered pigmented cells that are positive for melanocytic markers (Fig. 35.340).35–37 In one patient, an association with hypertrichosis was documented.38
• Granular cell neurofibroma is focally composed of tumor cells with abundant periodic acid-Schiff-positive diastase-resistant granular cytoplasm.3 Diagnosis is often dependent on identifying areas with more typical morphology.
• Epithelioid neurofibroma is focally composed of epithelioid cells with pink cytoplasm in a background of an otherwise typical neurofibroma.
• Dendritic cell neurofibroma with pseudorosettes is a distinctive variant of neurofibroma with a nodular growth pattern and two cell types.39 Small round, dark, lymphocyte-like dark cells surround larger cells with vesicular nuclei, frequent intranuclear inclusions and abundant pale cytoplasm, resulting in a distinctive pseudorosette appearance (Figs 35.341 and 35.342). Both cell types are positive for S100 protein and CD57.40 A case with a granulomatous. pattern has been reported.41 An
1793 Benign neural tumors
1794 Connective tissue tumors
intraneural example of this variant has recently been documented.42 An intraoral case has been described.43 Similar lesions may be seen in neurofibromatosis type I.44
• Cellular neurofibroma with atypia (atypical neurofibroma) refers to a sporadic neurofibroma with increased cellularity, focal atypia and very low mitotic activity.45,46 These lesions appear to have a benign behavior. Similar lesions, however, in the context of neurofibromatosis type I and in the presence of any mitotic activity, should be regarded as evidence of malignancy.
• Pacinian neurofibroma is best considered as a variant of schwannoma (see above). Examples of nerve sheath myxomas were formerly described as pacinian neurofibromas.
• Lipomatous neurofibroma refers to the presence of collections of mature fat cells within a neurofibroma.47–49 It seems to be more common in the head and neck.50 Distinction from a neurotized nevus with fatty metaplasia may be impossible (see differential diagnosis).
• Hybrid tumors show features of both neurofibroma and schwannoma; rarely features of perineurioma may be seen.51–53 Perineurial differentiation and intraneural occurrence have also been documented.54,55 Monosomy 22 has been identified in a subset of hybrid tumors.56 In addition, it has been shown that hybrid tumors are strongly associated with neurofibromatosis and schwannomatosis.57
• A single case with clear cell change, one with balloon cell change and one case with prominent sclerosis mimicking sclerotic fibroma have been reported.58–61 A variant described as angioneurofibroma has been documented.62
• Atypical neurofibromatous neoplasms of uncertain biologic potential (ANNUBP) is a term proposed for neurofibromas that were previously described inconsistently as atypical neurofibromas or low-grade malignant peripheral nerve sheath tumors.14,63 According to the consensus report nuclear atypia alone is not enough for a diagnosis of malignancy. However, tumors with cytologic atypia accompanied by loss of the usual neurofibroma architecture, high cellularity, and/ or mitotic activity higher than 1 per 50 HPF but less than 3 per 10 HPF are worrisome. It has therefore been proposed that when a tumor shows at least two of the above criteria the designation ‘neurofibromatous neoplasms of uncertain biologic potential’ should be used. Some of these tumors may show diminished S100 protein and SOX10 immunoreactivity, loss of p16/CDKN2A expression, elevated Ki67, and overexpression of TP53 as seen in malignant peripheral nerve sheath tumors. Also, as described in half of the latter, complete loss of trimethylated histone 3 lysine 27 expression, may be seen.14
• the central or acoustic form or type II neurofibromatosis (NF2; NF2 gene encoding merlin protein at 22q12.2). A third variant, segmental neurofibromatosis, has also been described. This occurs as a result of mosaicism in either NF1 or NF2, more commonly the former.
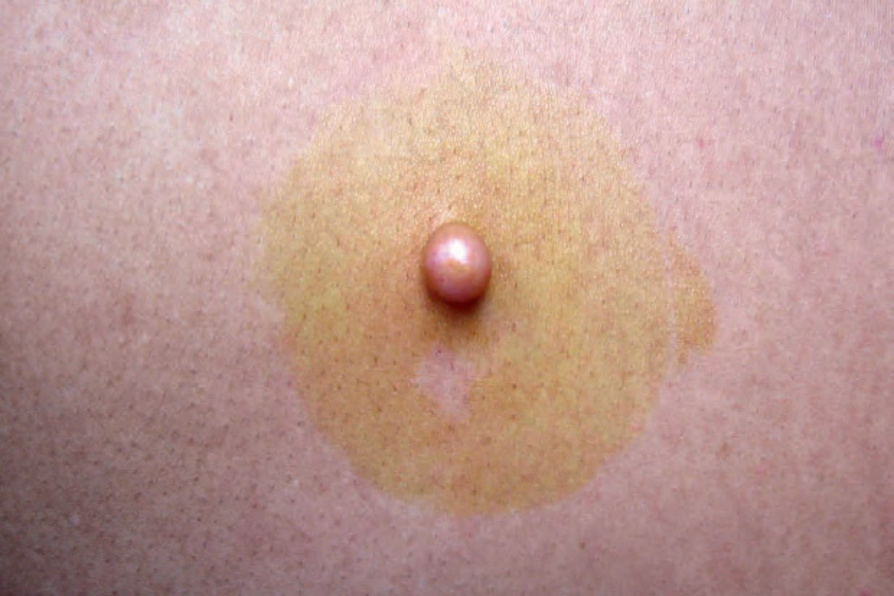
Fig. 35.324 Neurofibroma: this example presented as a circumscribed firm exophytic nodule. By courtesy of J. Dayrit, MD, Manila, The Philippines.
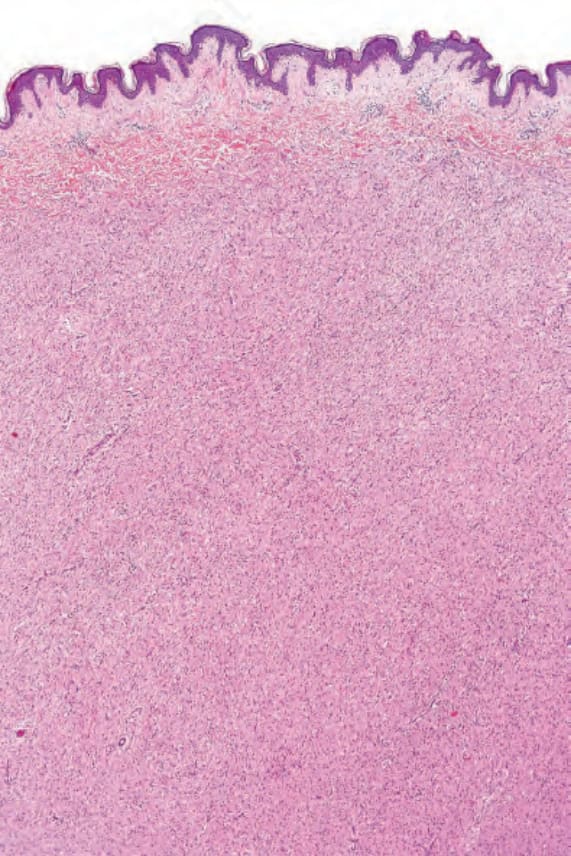
Fig. 35.325 Neurofibroma: the tumor is composed of small spindled cells with indistinct cell borders.
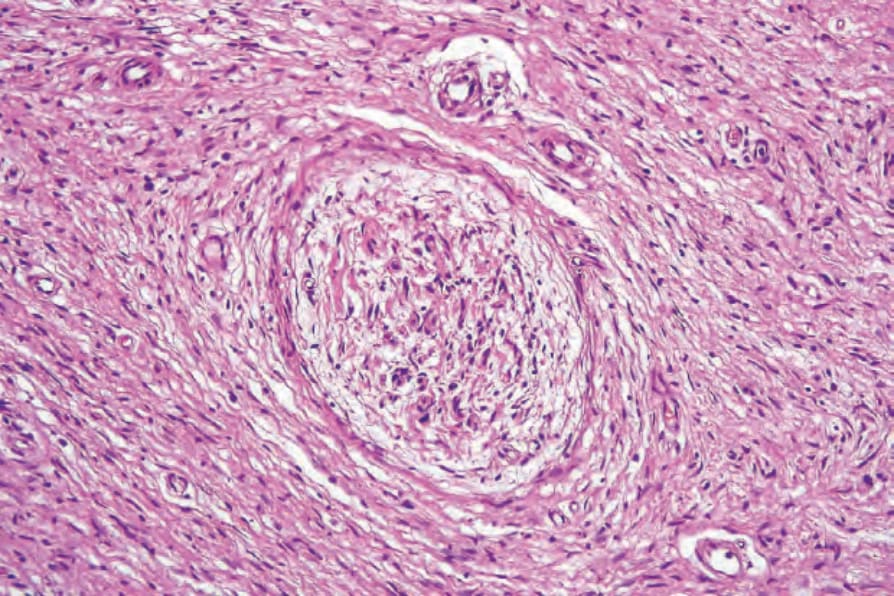
Fig. 35.326 Neurofibroma: a large nerve trunk is present in the center of the field.
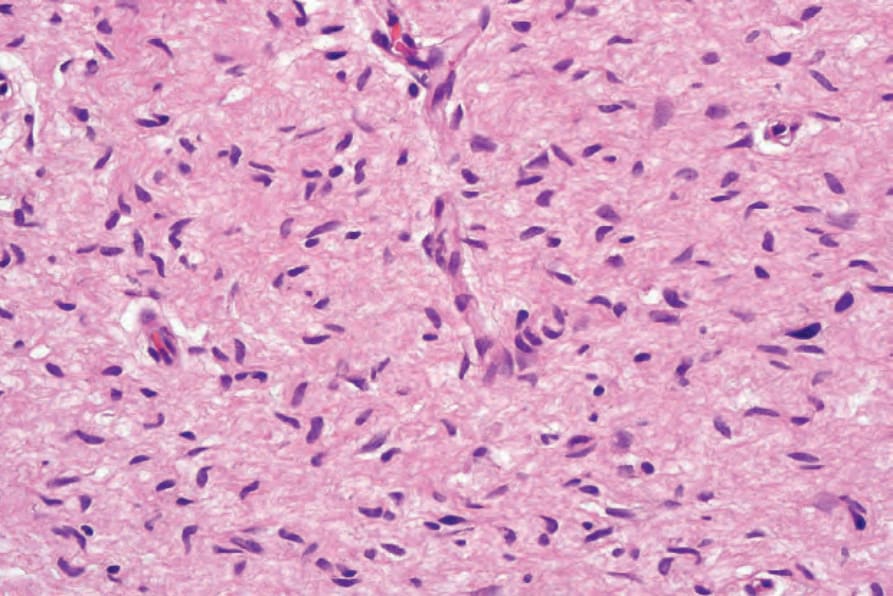
Fig. 35.327 Neurofibroma: the nuclei of the spindled cells are characteristically elongated and wavy.
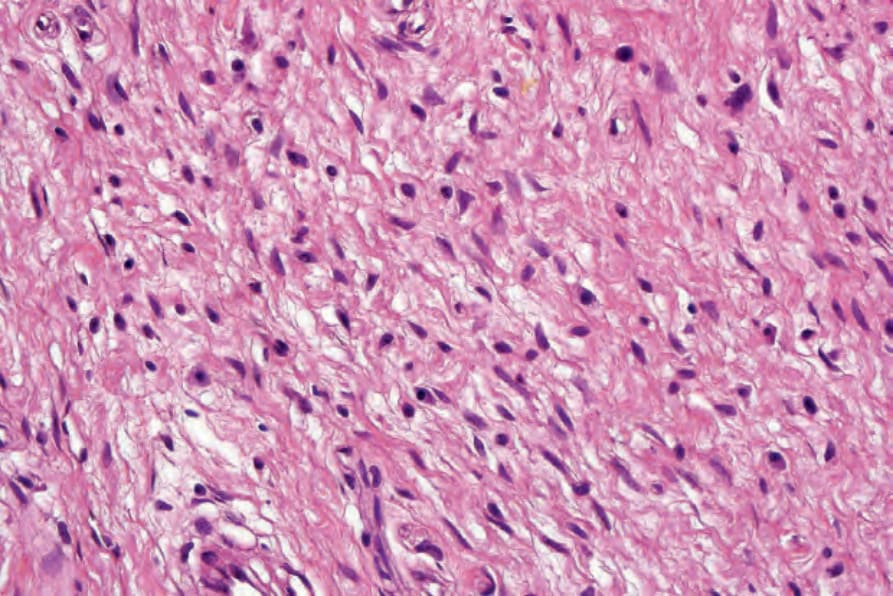
Fig. 35.328 Neurofibroma: mast cells with granular eosinophilic cytoplasm are frequently seen in these tumors.
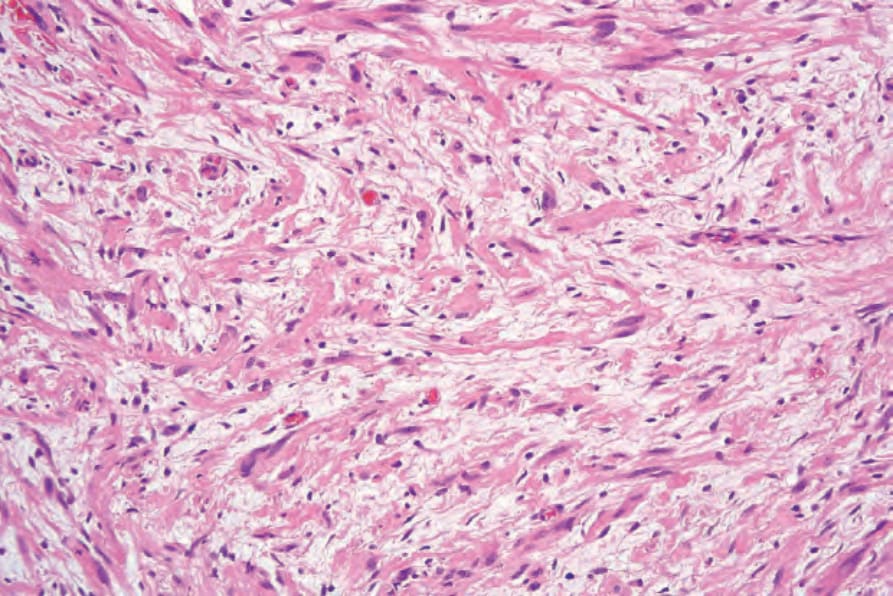
Fig. 35.329 Neurofibroma: the collagen content is highly variable, but may be prominent, as in this example.
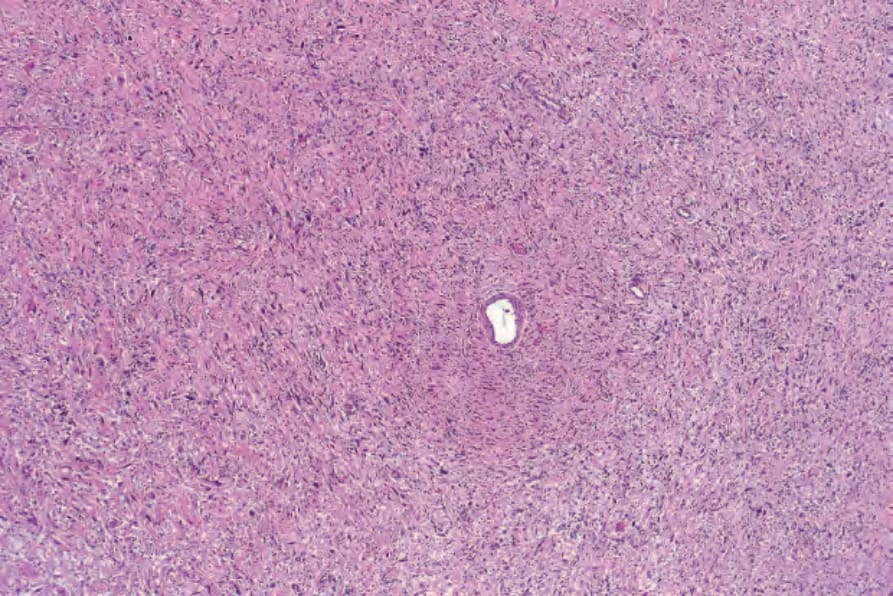
Fig. 35.330 Atypical neurofibroma: even at this magnification, hyperchromatic and enlarged nuclei are evident.
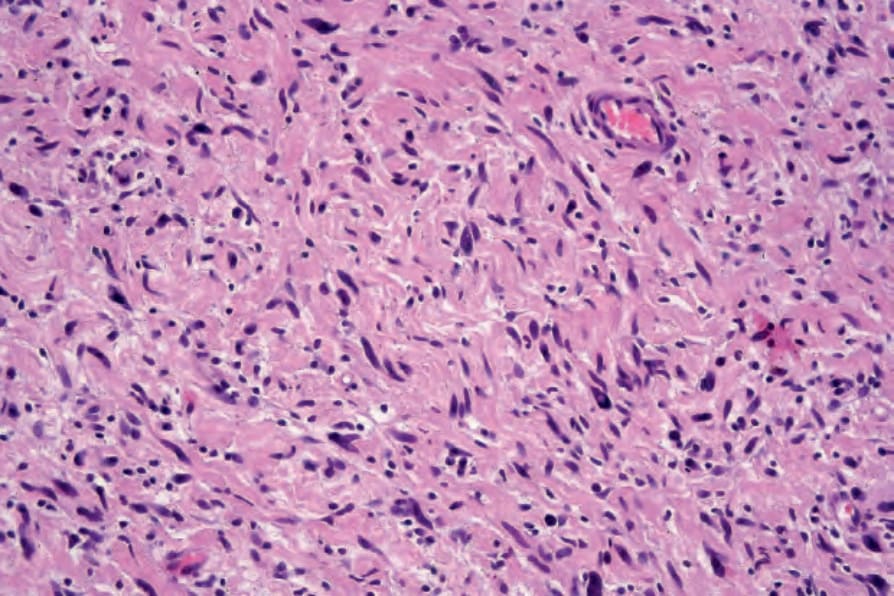
Fig. 35.331 Atypical neurofibroma: high-power view showing pleomorphic nuclei.
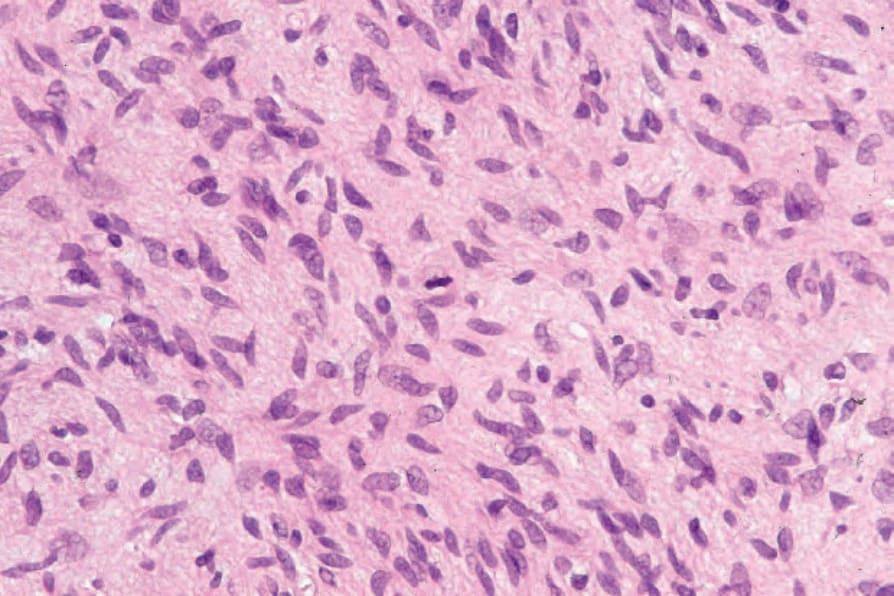
Fig. 35.332 Atypical neurofibroma: very occasionally, single mitoses may be identified.
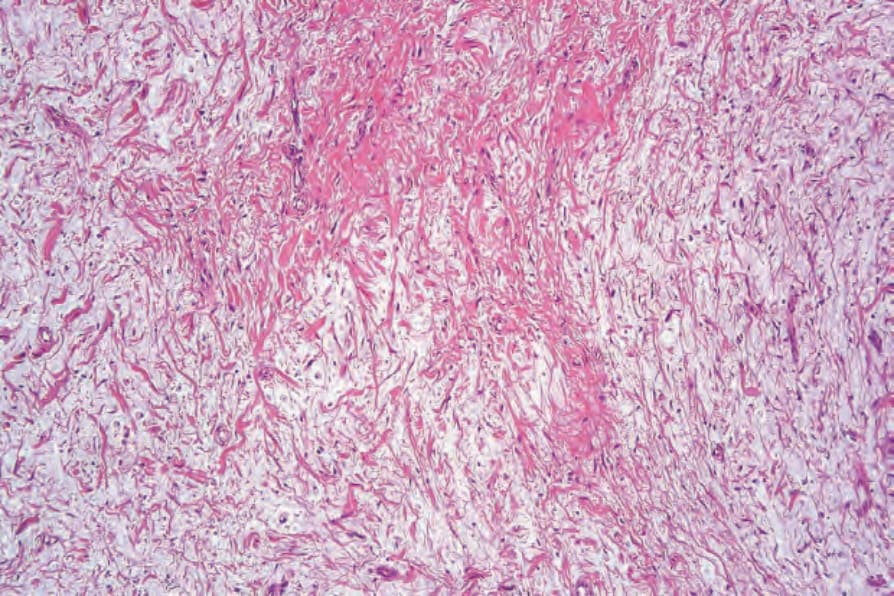
Fig. 35.333 Myxoid neurofibroma: in this variant, there is marked stromal mucin.
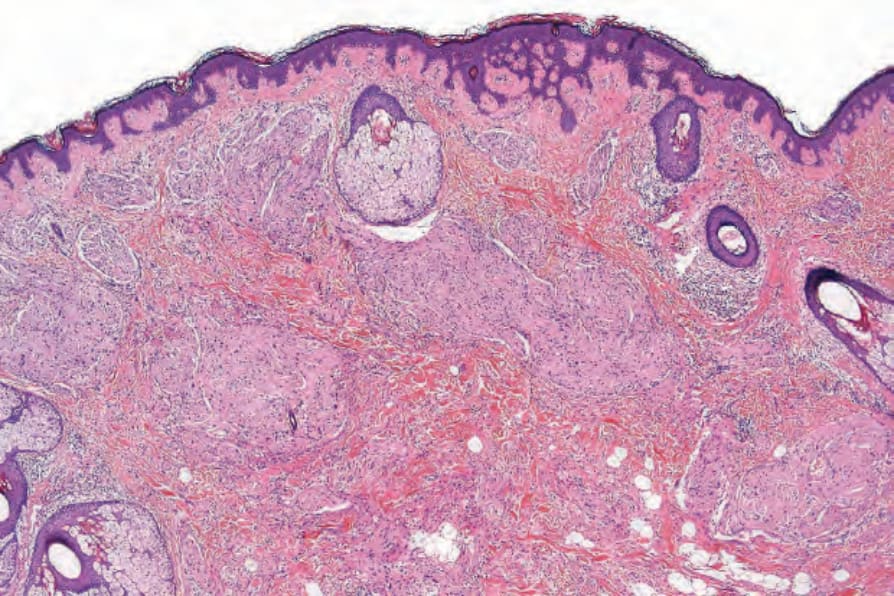
Fig. 35.334 Plexiform neurofibroma: thickened, haphazardly distributed nerve trunks are present in the reticular dermis.
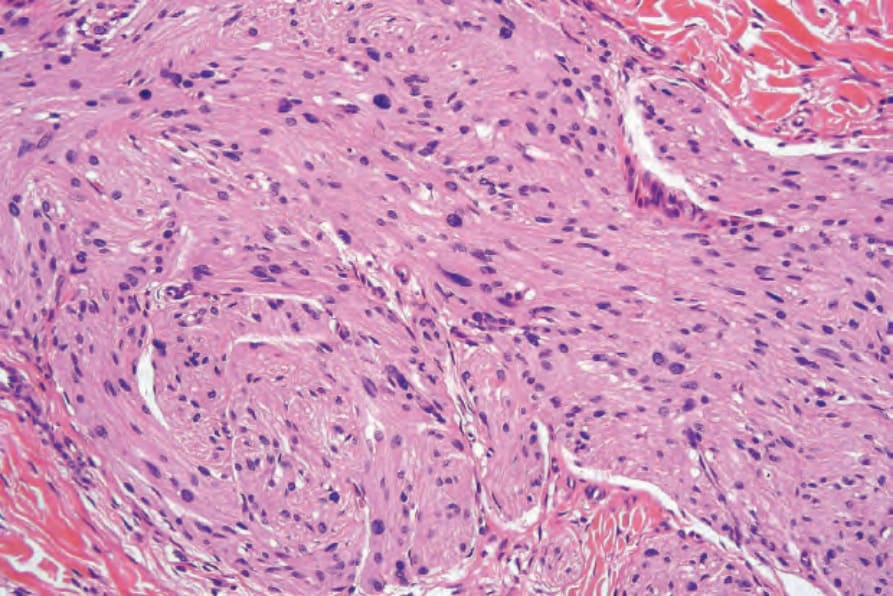
Fig. 35.335 Plexiform neurofibroma: hypertrophied nerves are seen embedded in a matrix of fibroblasts and Schwann cells.
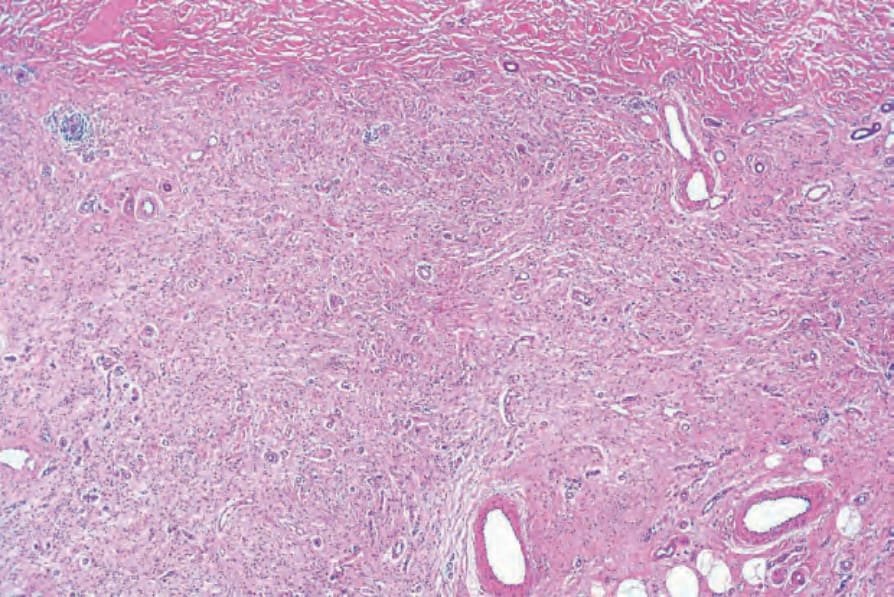
Fig. 35.336 Diffuse neurofibroma: both the papillary and the reticular dermis are extensively infiltrated by neurofibromatous tissue.
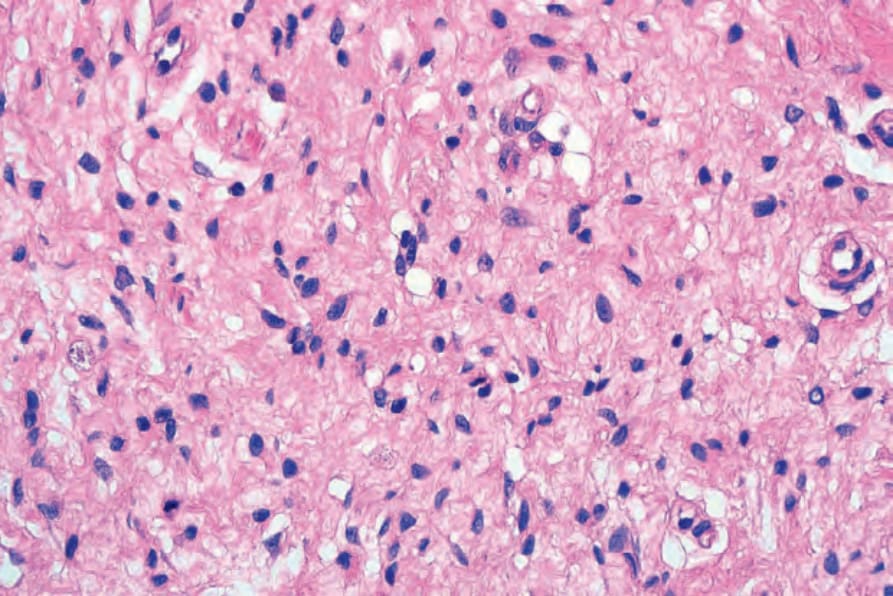
Fig. 35.337 Diffuse neurofibroma: high-power view.
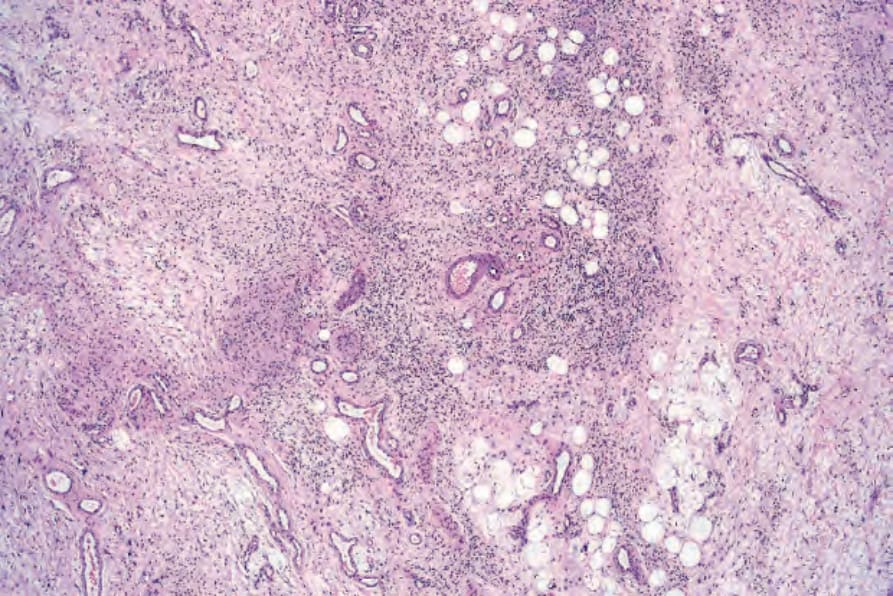
Fig. 35.338 Diffuse neurofibroma: there is marked organoid differentiation.
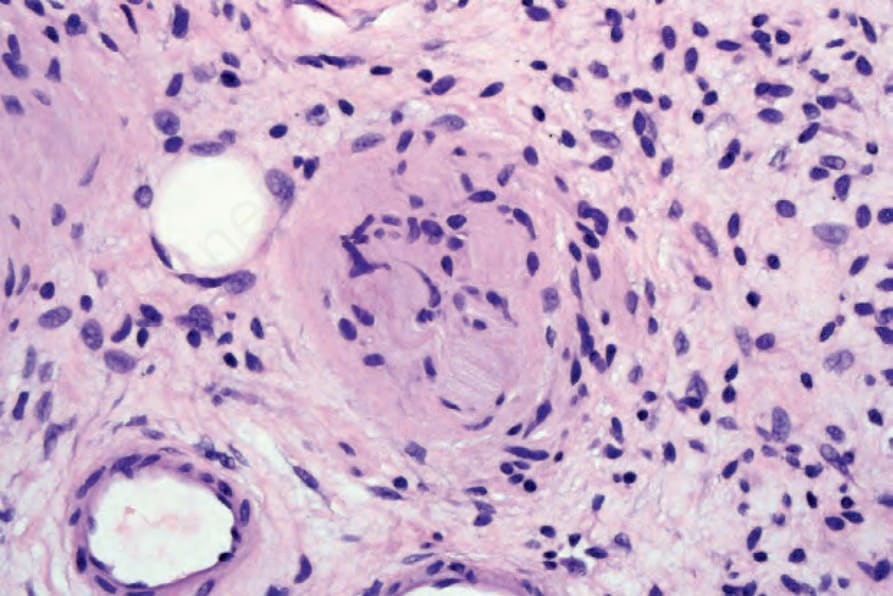
Fig. 35.339 Diffuse neurofibroma: in the center of the field there is differentiation towards a Meissner’s corpuscle.
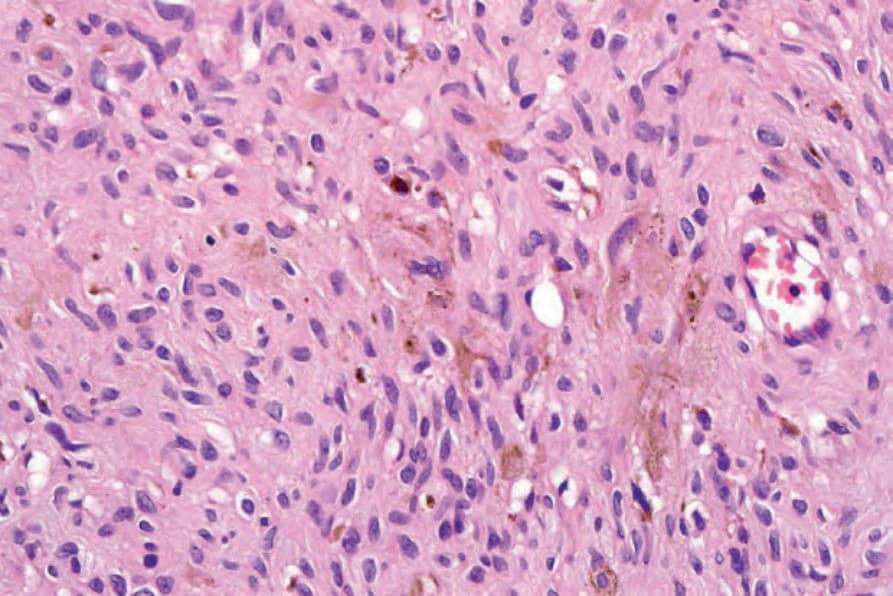
Fig. 35.340 Pigmented neurofibroma: note the pigmented dendritic cells. By courtesy of H. Diwan, MD, Houston, Texas, USA.
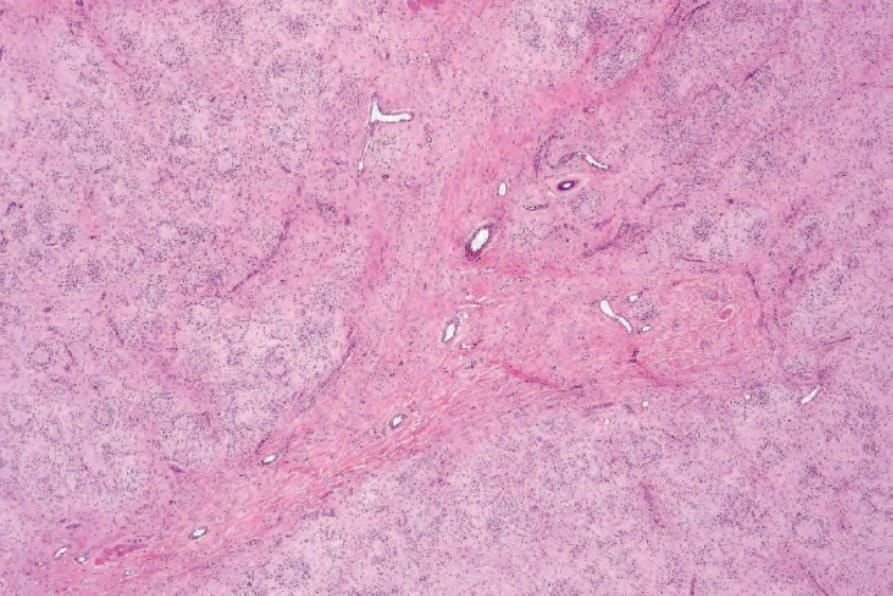
Fig. 35.341 Neurofibroma with pseudorosettes: low-power view showing conspicuous pseudorosettes.
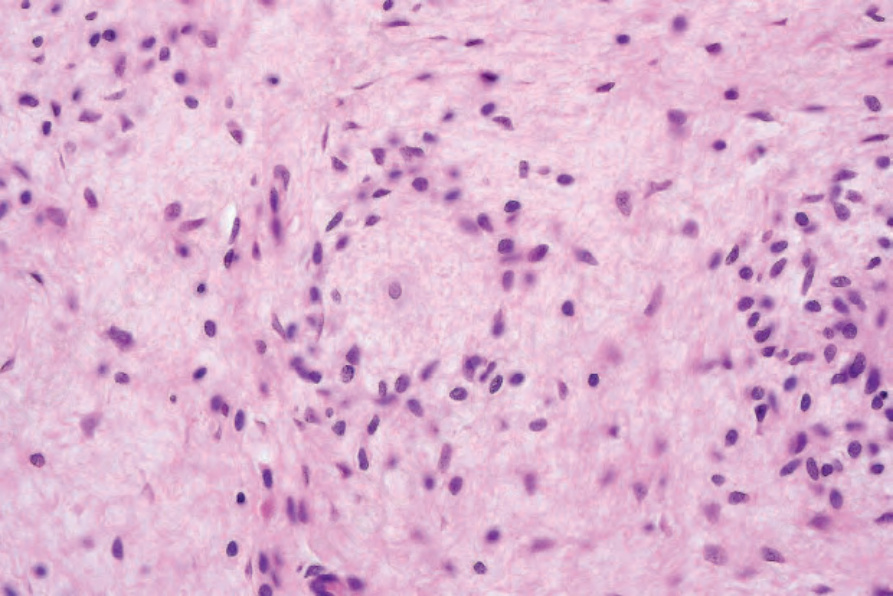
Fig. 35.342 Neurofibroma with pseudorosettes: there is a single central large pale cell surrounded by a mantle of small lymphocyte-like cells.
Other variants of neurofibromatosis have been documented and include hereditary spinal neurofibromatosis, schwannomatosis, familial intestinal neurofibromatosis, autosomal dominant ‘café-au-lait spots alone,’ autosomal dominant ‘neurofibromas alone,’ Watson syndrome, Noonan/ neurofibromatosis syndrome and multiple nevi, multiple schwannomas, and multiple vaginal leiomyomas.1,2
Neurofibromatosis type I is an important congenital neurocutaneous disorder with an autosomal dominant mode of inheritance, affecting about 1/3000 live births; a number of cases also arise as a result of spontaneous germline mutation.3–12 It encompasses a constellation of signs and symptoms and may involve most systems of the body. The gene for NF1 has been cloned to chromosome 17q11.2.13–16 The encoded protein is called neurofibromin, which helps control Ras activity in cells.17–19 Tumors are generated with loss of heterozygosity at the NF1 wild type allele. Chromosomal imbalances are more common in NF1-associated neurofibromas than in sporadic neurofibromas.20 Other chromosomal imbalances described in NF1 include losses in chromosomes 19 and 22q.20 See also the neurofibroma section above.
Patients with NF1 and no evidence of cutaneous neurofibromas have a 3-bp inframe deletion in exon 17 of the NF1 gene.21
Recently, a neurofibromatosis type 1-like autosomal dominant syndrome lacking NF1 mutations but associated with germline inactivating mutations in SPRED1 (sprout-related EVH1 domain-containing protein 1) has been described.22 In this syndrome, patients present with café-au-lait macules, axillary freckling and macrocephaly.
The National Institutes of Health (NIH) criteria for the diagnosis of NFI include two or more of the following:23
• six or more café-au-lait macules with a diameter of greater than 5.0 mm in children less than 6 years of age and greater than 15 mm in older individuals,
• two or more neurofibromas of any type or one plexiform neurofibroma,
• freckling in the axillary or inguinal regions,
• an optic nerve glioma,
• two or more Lisch nodules (iris hamartomas),
• a distinctive osseous lesion, such as dysplasia of the sphenoid bone or thinning of the cortex of long bones, with or without pseudoarthrosis,
• a first-degree relative with NF1. Neurofibromatosis type I shows very wide clinical variability. A large study has demonstrated an association between several pairs of features in affected probands:
• intertriginous freckling and Lisch nodules,
• discrete neurofibromas and plexiform neurofibromas,
• discrete neurofibromas and Lisch nodules,
• plexiform neurofibromas and scoliosis,
• learning disability or mental retardation and seizures.24
Differential diagnosis Small biopsies of plaque-stage dermatofibrosarcoma protuberans may be difficult to distinguish from a neurofibroma. The former, however, has a distinctive lace-like pattern of infiltration of the fat, the dermal bundles tend to be parallel to the epidermis and tumor cells are negative for S100 protein and positive for CD34. Old neurotized nevi are often indistinguishable from neurofibroma if no residual nevus cells or epidermal component are present. S100 protein will be positive in both lesions, but neurotized nevi tend to be symmetrical and are also more often NSE positive. Importantly, expression of melanocytic markers may be seen in neurofibromas (including the epithelioid variant) arising in patients with neurofibromatosis.64
Café-au-lait macules are flat, light-brown lesions which may be distributed anywhere on the integument, but are found predominantly on unexposed surfaces of the body (Fig. 35.343). Up to 10% of the population may have solitary lesions from birth. Patients may also exhibit more darkly colored macules overlying cutaneous plexiform neurofibromas (Fig. 35.344). It has been suggested that dermal fibroblast-derived stem cell factor and hepatocyte growth factor may play a role in the development of the hyperpigmentation.25
The use of CD34 as a tool for differentiating neurofibromas (characteristic fingerprint pattern seen in neurofibroma as opposed to melanoma) from spindle cell melanomas has been reported but it is not an entirely reliable feature.65–67
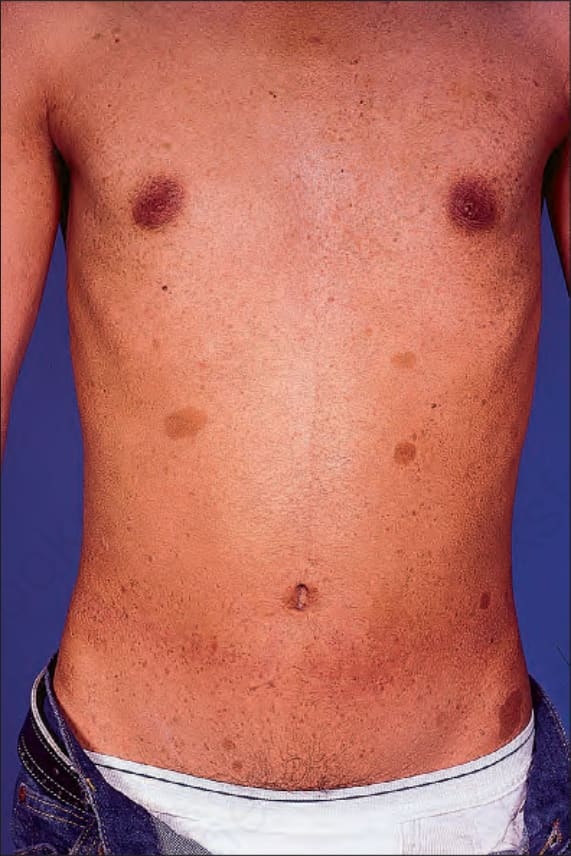
Fig. 35.343 Type I neurofibromatosis: the presence of typical café-au-lait macules is characteristic. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
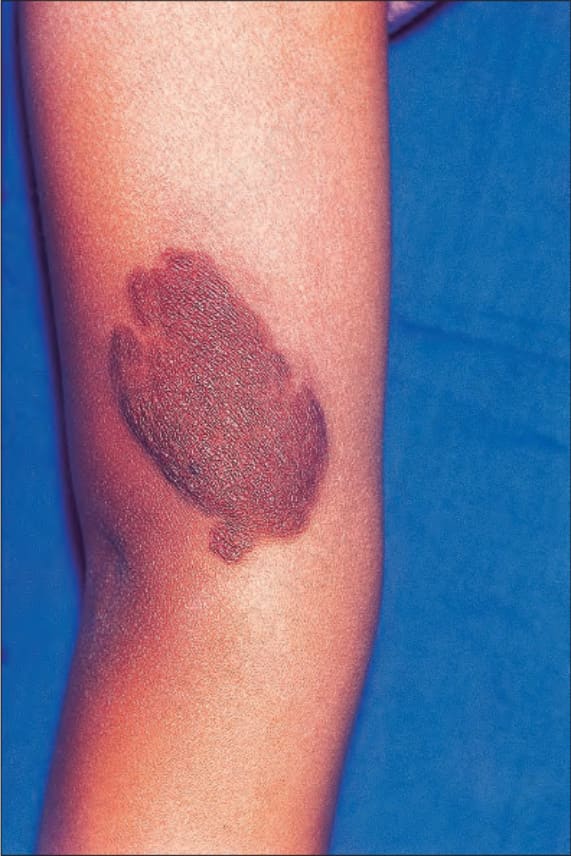
Fig. 35.344 Type I neurofibromatosis: this heavily pigmented raised lesion overlies a cutaneous plexiform neurofibroma. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.