疾病定義與分類
紅斑性狼瘡 (lupus erythematosus) 是一種複雜的疾病,伴隨眾多臨床徵象與症狀,以及範圍極廣的實驗室異常。它呈現出不同預後的疾病譜:一端為良性、僅侷限於皮膚的變異型(侷限型 discoid),另一端則可能是致命的全身性疾病。各亞型如 Table 17.1 所示。
雖然確切病因不明,但一般認為遺傳因素、自體抗體、免疫複合體、荷爾蒙與其他因子之間的交互作用,是疾病發生的原因。有證據顯示全身性變異型的發生率正在上升,但由於診斷更早、治療更有效,死亡率已顯著下降,成人 10 年整體存活率目前已超過 90%。然而,腎臟與/或神經系統侵犯的存在,仍是不良預後的指標。
兒童全身性紅斑性狼瘡 (systemic lupus erythematosus, SLE) 是一種侵襲性疾病,死亡率相當高,主要源於腎臟疾病的發生率。即使使用 corticosteroid 與免疫抑制治療,死亡率仍高達 15%。

表 17-1:紅斑性狼瘡的亞型 (lupus erythematosus: subtypes)。
Table 17.1 Lupus erythematosus: subtypes
紅斑性狼瘡的亞型範圍包括:侷限型盤狀紅斑性狼瘡 (discoid lupus erythematosus, localized)、泛發型盤狀紅斑性狼瘡 (discoid lupus erythematosus, generalized)、疣狀紅斑性狼瘡 (verrucous lupus erythematosus)、凍瘡型紅斑性狼瘡 (chilblain lupus erythematosus)、伴有盤狀紅斑性狼瘡樣皮膚病的慢性肉芽腫病 (chronic granulomatous disease with discoid lupus erythematosus-like dermatosis)、紅斑性狼瘡-多形性紅斑症候群 (lupus erythematosus-erythema multiforme syndrome)、亞急性皮膚紅斑性狼瘡 (subacute cutaneous lupus erythematosus)、深部紅斑性狼瘡 (lupus erythematosus profundus)、全身性紅斑性狼瘡 (systemic lupus erythematosus)、藥物誘發型紅斑性狼瘡 (drug-induced lupus erythematosus)、C2 缺乏性紅斑性狼瘡樣症候群 (C2 deficiency lupus erythematosus-like syndrome),以及新生兒紅斑性狼瘡 (neonatal lupus erythematosus)。
臨床特徵 (Clinical Features)
盤狀紅斑性狼瘡 (Discoid lupus erythematosus)
盤狀紅斑性狼瘡 (discoid lupus erythematosus, DLE) 是最常見的型態,可細分為侷限型 (localized) 與泛發型 (generalized) 變異型。此區分具有預後意義,因為侷限型 DLE 病人中僅約 1% 會發展為全身性疾病,但泛發型者中約有 5%(特別是那些持續性貧血、白血球低下、血小板低下、偽陽性 Wassermann 反應,以及高效價抗核因子者)會發展為完全表現的 SLE。多達 20% 的全身性狼瘡病人會出現盤狀病灶。甲周毛細血管擴張 (periungual telangiectasia)、硬指 (sclerodactyly),以及 Raynaud 現象 (Raynaud phenomenon) 的存在,也可能預示潛在的疾病進展。對 DLE 病人不應過度誇大其發展為全身性侵犯的風險,因為此風險並不高。
DLE 曾被描述與下列情況相關:骨斑點病 (osteopoikilosis)、α1-antitrypsin 缺乏、結節性多動脈炎 (polyarteritis nodosa)、Addison 病、慢性肉芽腫病 (chronic granulomatous disease)、營養不良性皮膚鈣質沉著 (dystrophic calcinosis cutis)、過敏性接觸性皮膚炎 (allergic contact dermatitis)、後天性部分脂肪萎縮 (acquired partial lipoatrophy)、作為對刺青的反應,以及在一位合併硬皮病 (scleroderma)、深部線狀硬斑病 (deep linear morphea) 與慢性 C 型肝炎病毒感染的病人身上。例外情況下,侷限型 DLE 曾被報告出現在先前受帶狀疱疹 (herpes zoster) 侵犯的部位,以及先前外傷的部位。
DLE 為持續性疾病,女性受影響的人數是男性的兩倍。雖然任何年齡層都可能受影響,但最常見於第三、第四與第五個十年(30–50 多歲),發生率高峰在 30 歲後期。兒童期表現者罕見。病灶通常出現在日曬部位,病人常經歷光敏性 (photosensitivity);可能在春夏季加重。在侷限型,通常侵犯頭部與頸部;但在泛發型變異型,病灶也可能出現在手臂、手部與手指的背側面,以及頸部的「V」形區域。非曝曬部位,包括軀幹、上肢,以及手掌與足底,也常受侵犯。
顏面斑塊最常出現在臉頰 (Fig. 17.1)。其他受侵犯部位包括鼻樑、耳朵、頸部與頭皮 (Figs 17.2–17.6)。頭皮相關的瘢痕化會繼之以永久性禿髮 (Figs 17.7 and 17.8)。頭皮侵犯在年輕時罹病的病人中較為常見,呈慢性病程,並與長期嚴重的疾病相關。眼部侵犯主要涵蓋眼瞼,其次為眼眶與角膜,其中眼瞼炎 (blepharitis) 是最常見的症狀。
早期病灶呈水腫性與紅斑性。已成形的 DLE 斑塊(直徑可達 10 cm 之多)通常覆有黏附性鱗屑,並伴隨表皮萎縮、毛囊擴張與栓塞 (Figs 17.9 and 17.10)。若移除鱗屑,常可在其底面看見附著的角質栓(「地毯釘」徵象,‘carpet tacks’ sign)。毛細血管擴張 (telangiectasia) 是常見的發現,病灶癒合時伴有瘢痕,且往往相當明顯。在深膚色或黑膚色個體中,斑塊可能呈色素減少或色素增加,這可能造成特別嚴重的毀容 (Figs 17.11–17.14)。
口腔侵犯發生於 20% 至 25% 的 DLE 病人,特別侵犯下唇的唇紅緣 (vermilion border)、齒槽突 (alveolar processes)、唇黏膜與頰黏膜 (Figs 17.15–17.17)。慢性病灶典型上呈紅斑性與萎縮性,帶有扇貝狀的白色角化邊緣與鄰近的毛細血管擴張。糜爛與潰瘍是額外的特徵。病灶有時與萎縮型扁平苔癬 (atrophic lichen planus) 難以區分。舌部侵犯表現為紅斑、龜裂以及乳頭萎縮。慢性狼瘡唇炎 (chronic lupus cheilitis) 與瘢痕性結瘢以及鱗狀細胞癌 (squamous carcinoma) 風險增加相關。生殖器皮膚與肛周黏膜侵犯偶有記載。
在侷限型 DLE 病人中,通常無全身性症狀。在泛發型,皮膚斑塊可能相當廣泛,少數病人可能出現 Raynaud 現象與關節痛 (Figs 17.23–17.25)。實驗室異常在泛發型較侷限型更常見。侷限型 DLE 不會出現貧血,但有時是泛發型變異型的特徵。白血球低下、紅血球沉降速率 (erythrocyte sedimentation rate, ESR) 上升,以及高γ球蛋白血症 (hypergammaglobulinemia) 在兩型皆可發生。抗核因子(瀰漫型 diffuse pattern)、Wassermann 反應陽性與類風濕因子 (rheumatoid factor) 也可能是特徵。抗雙股 DNA (anti-double-stranded DNA, dsDNA) 抗體在少數播散型 DLE 病人中可被偵測到,且這些病人常轉變為全身性疾病;偶爾會出現抗單股 DNA (single-stranded DNA, ssDNA) 抗體。尿液分析與腎功能檢查正常。
疣狀(肥厚型)盤狀紅斑性狼瘡 (Verrucous [hypertrophic] discoid lupus erythematosus)
疣狀(肥厚型)DLE 表現為疣狀、過度角化的丘疹與斑塊,好發於臉部、頭皮、唇部黏膜以及上肢 (Figs 17.18 and 17.19)。它影響約 2% 的慢性盤狀疾病病人。雖然疣狀(肥厚型)過度角化的皮膚變化曾在 SLE 病人身上被觀察到,但這類案例屬例外。有時手掌與足底受侵犯,偶爾甲部也受侵犯 (Figs 17.20 and 17.21)。罕見的表現包括手臂與手部出現結節性角化棘皮瘤樣 (keratoacanthoma-like)、鱗狀細胞癌樣 (squamous cell carcinoma-like) 與肥厚型扁平苔癬樣 (hypertrophic lichen planus-like) 的病灶 (Fig. 17.22)。一種伴隨皮下組織壞死的變異型稱為肥厚兼深部紅斑性狼瘡 (lupus erythematosus hypertrophicus et profundus)。
皮膚鱗狀細胞癌 (cutaneous squamous cell carcinoma),以及較少見的基底細胞癌 (basal cell carcinoma),可發生於慢性病灶(包括肥厚型變異型)的病人。鱗狀細胞癌主要發生於男性,特別侵犯頭皮,有時伴隨早期轉移。最近曾報告一例在 DLE 瘢痕中發生的非典型纖維黃色瘤 (atypical fibroxanthoma)。
腫脹性紅斑性狼瘡 (Lupus erythematosus tumidus)
腫脹性紅斑性狼瘡 (lupus erythematosus tumidus) 是皮膚狼瘡中一個獨特的亞型,臨床上主要出現於 DLE 病人,極少出現於 SLE 病人。其特徵為紅斑性丘疹、斑塊甚至結節,呈蕁麻疹樣外觀,主要出現於臉部、頸部與軀幹的日曬皮膚。不會發生瘢痕。此狼瘡變異型於第 8 章有更詳細的討論。
凍瘡型紅斑性狼瘡 (Chilblain lupus erythematosus)
凍瘡型紅斑性狼瘡 (chilblain lupus erythematosus, CHLE) 約占 DLE 病例的 11%,在冬季或暴露於寒冷、潮濕與陰冷的環境之後發病。雖然 CHLE 最常出現於散發性病人,但近來也有家族性發生的報告。家族性 CHLE 病人在位於染色體 3p、編碼一種 DNA 專一性 3′-5′ 外切核酸酶 1 (3′-5′ exonuclease 1) 的 TREX1 基因上具有突變。此外,曾在單一家族報告一個 SAMHD1(SAM domain and HD domain-containing protein 1,一種降解去氧核苷酸的磷酸水解酶)的突變。這些病人已被證實為自體顯性遺傳 (autosomal dominant inheritance)。家族性 CHLE 一般於兒童早期表現,散發性 CHLE 則通常影響中年女性。病人在手指、足跟與足底發展出搔癢、疼痛、丘疹紅斑性或藍紫色的斑塊與結節;手部、小腿、膝部、指關節、肘部、鼻部與耳朵受侵犯較少 (Fig. 17.26)。有時也會出現過度角化龜裂的病灶與潰瘍。CHLE 可能以類似白斑 (vitiligo) 的脫色表現。雖然病人通常在典型盤狀皮疹出現許多年後才發展出凍瘡,但病灶可能同時出現,有時凍瘡是唯一的表現。部分散發性 CHLE 病人合併有冷纖維蛋白原血症 (cryofibrinogenemia) 或冷凝集素 (cold agglutinin)。約 20% 的散發性 CHLE 病人會發展為 SLE,特別是那些同時發展出盤狀與凍瘡樣病灶者,以及除凍瘡外還有 DLE-多形性紅斑樣症候群者。家族性 CHLE 病人中無人進展為 SLE。
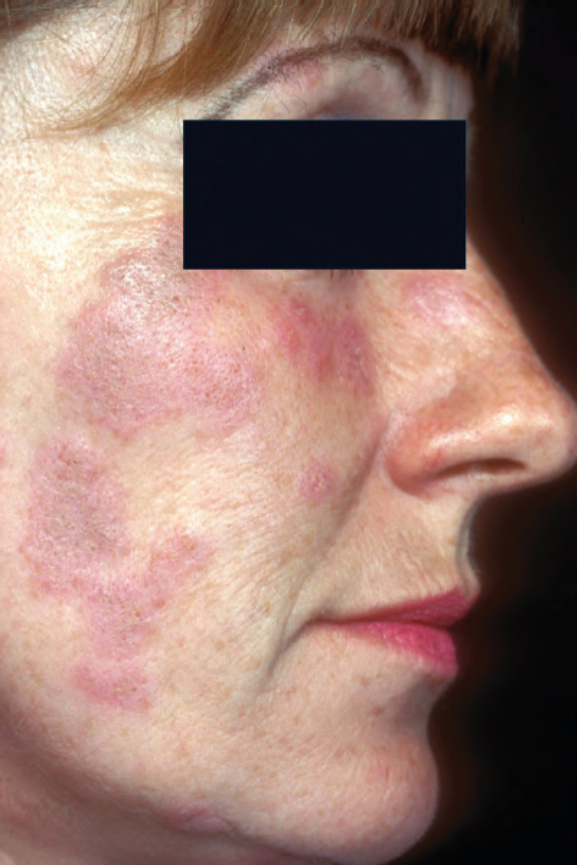
圖 17-1:盤狀紅斑性狼瘡——一位女性病人臉頰上出現典型斑塊。注意紅斑與鱗屑。這是一個特徵性部位。
Fig. 17.1 Discoid lupus erythematosus: typical plaques are present on the cheek of a female. Note the erythema and scale. This is a characteristic site. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
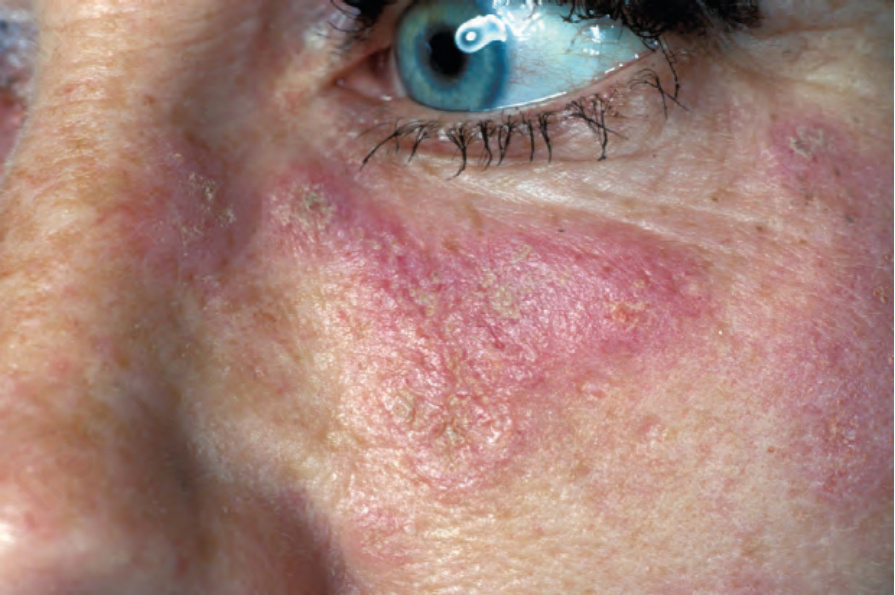
圖 17-2:盤狀紅斑性狼瘡——特寫顯示紅斑與鱗屑。
Fig. 17.2 Discoid lupus erythematosus: close-up view showing erythema and scale. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
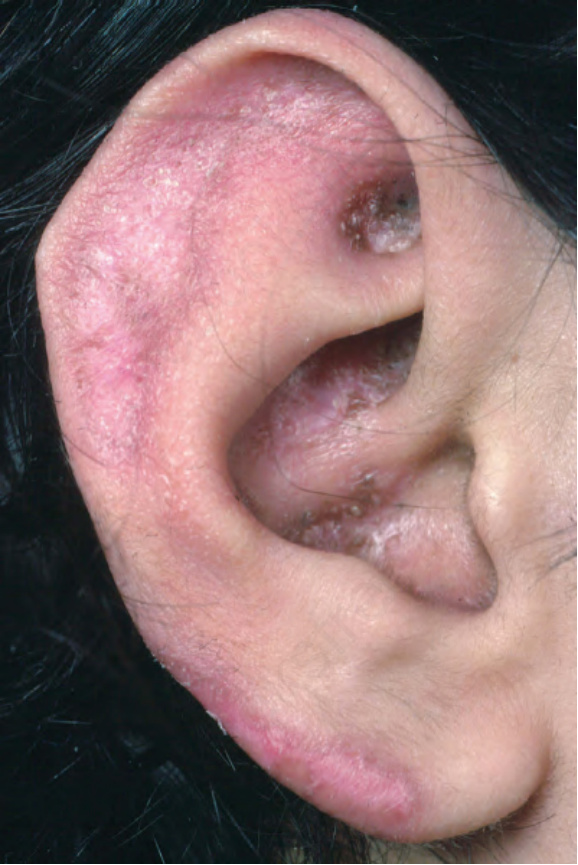
圖 17-3:盤狀紅斑性狼瘡——耳垂上有鱗屑與瘢痕,這是常受侵犯的部位。
Fig. 17.3 Discoid lupus erythematosus: there is scaling and scarring on the ear lobe, a commonly affected site. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
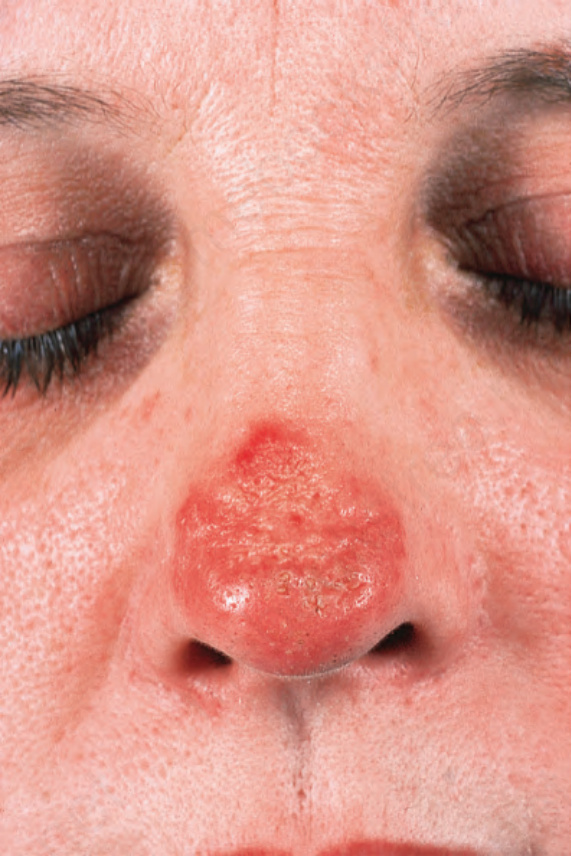
圖 17-4:盤狀紅斑性狼瘡——這位嚴重受侵犯的病人顯示已癒合的潰瘍,伴有明顯瘢痕與毀容。
Fig. 17.4 Discoid lupus erythematosus: this severely affected patient shows healed ulceration with marked scarring and disfigurement. By courtesy of the Institute of Dermatology, London, UK.
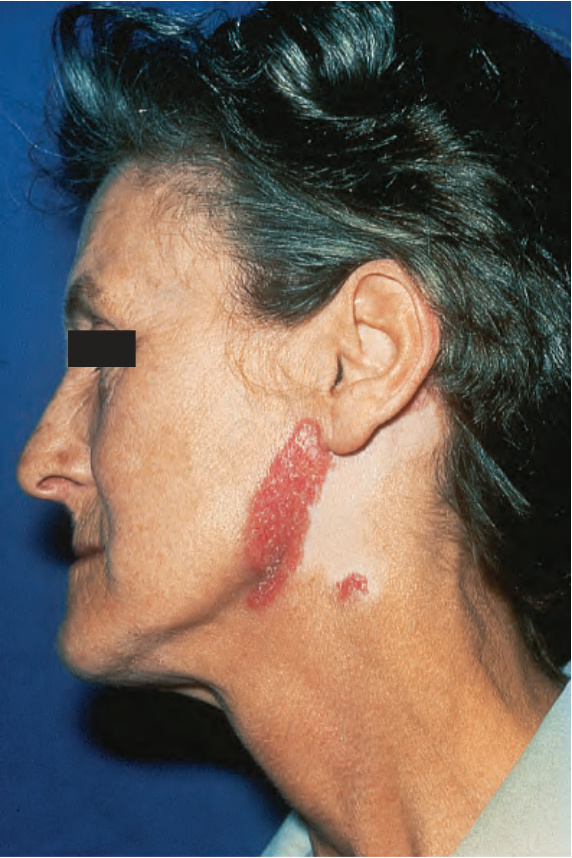
圖 17-5:盤狀紅斑性狼瘡——此慢性病灶中有明顯瘢痕。
Fig. 17.5 Discoid lupus erythematosus: in this chronic lesion there is marked scarring. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
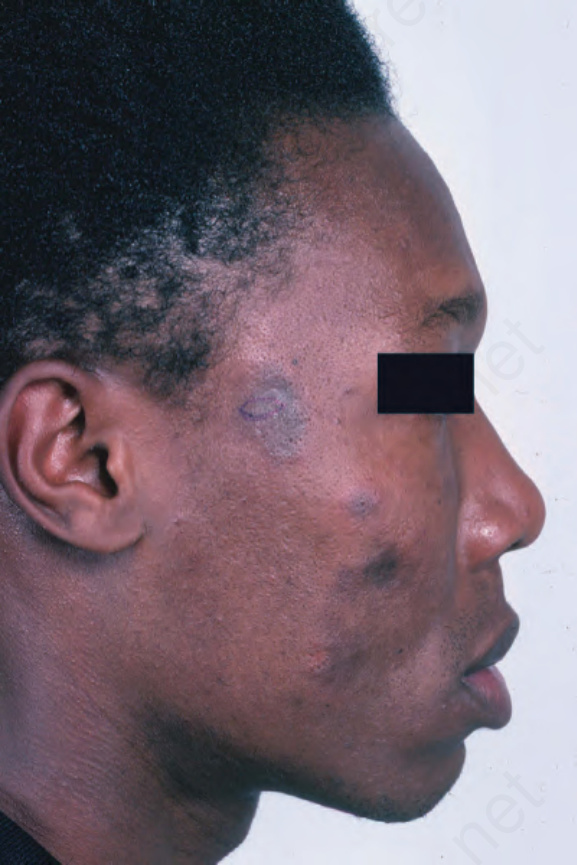
圖 17-6:盤狀紅斑性狼瘡——深膚色人種常受侵犯。
Fig. 17.6 Discoid lupus erythematosus: dark-skinned races are commonly affected. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
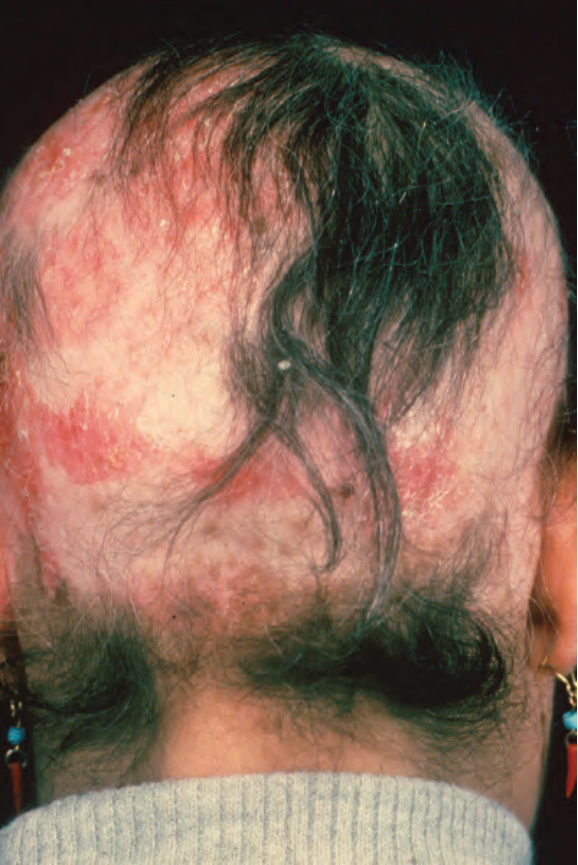
圖 17-7:盤狀紅斑性狼瘡——掉髮是永久性的。
Fig. 17.7 Discoid lupus erythematosus: hair loss is permanent. By courtesy of the Institute of Dermatology, London, UK.
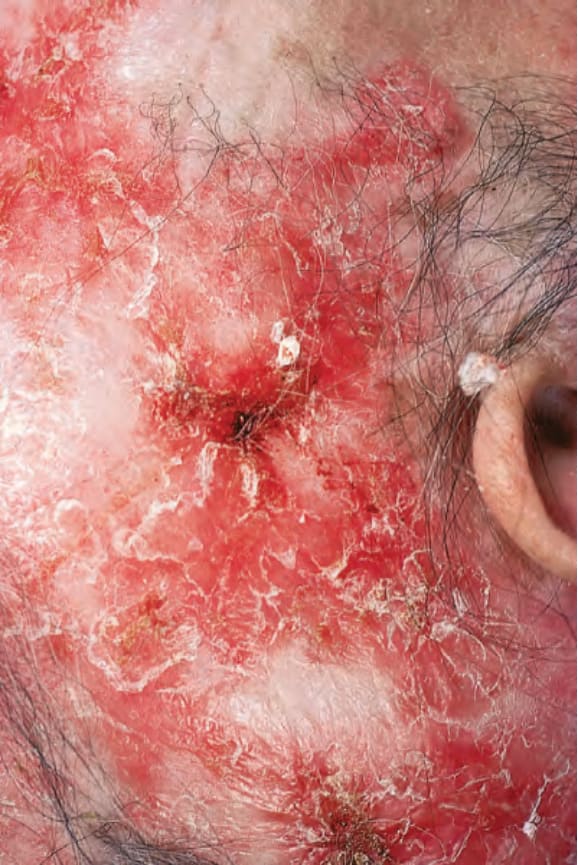
圖 17-8:盤狀紅斑性狼瘡——有禿髮並伴隨非常明顯的瘢痕。
Fig. 17.8 Discoid lupus erythematosus: there is alopecia with very marked scarring. By courtesy of the Institute of Dermatology, London, UK.
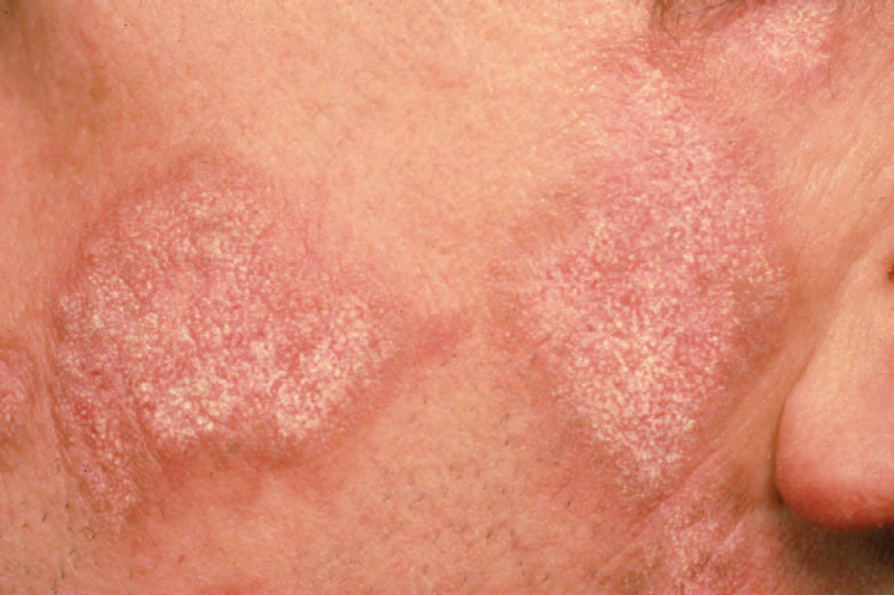
圖 17-9:盤狀紅斑性狼瘡——鱗屑特寫。注意紅斑性邊緣。
Fig. 17.9 Discoid lupus erythematosus: close-up view of scale. Note the erythematous border. By courtesy of the Institute of Dermatology, London, UK.
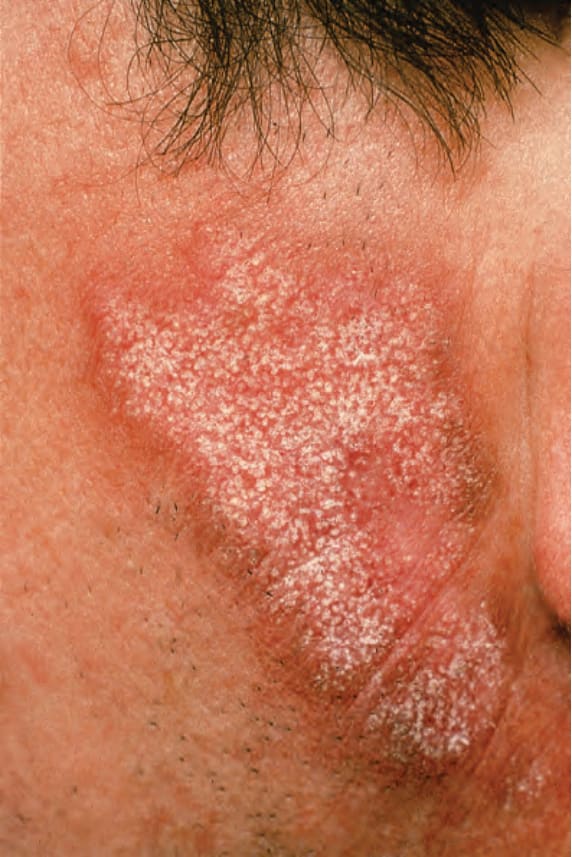
圖 17-10:盤狀紅斑性狼瘡——特寫顯示毛囊栓塞 (follicular plugging)。
Fig. 17.10 Discoid lupus erythematosus: close-up view showing follicular plugging. By courtesy of the Institute of Dermatology, London, UK.
圖 17-11:盤狀紅斑性狼瘡——在深膚色病人中,色素增加病灶可造成非常嚴重的毀容。
Fig. 17.11 Discoid lupus erythematosus: foci of hyperpigmentation can be very disfiguring in dark-skinned patients. By courtesy of the Institute of Dermatology, London, UK.
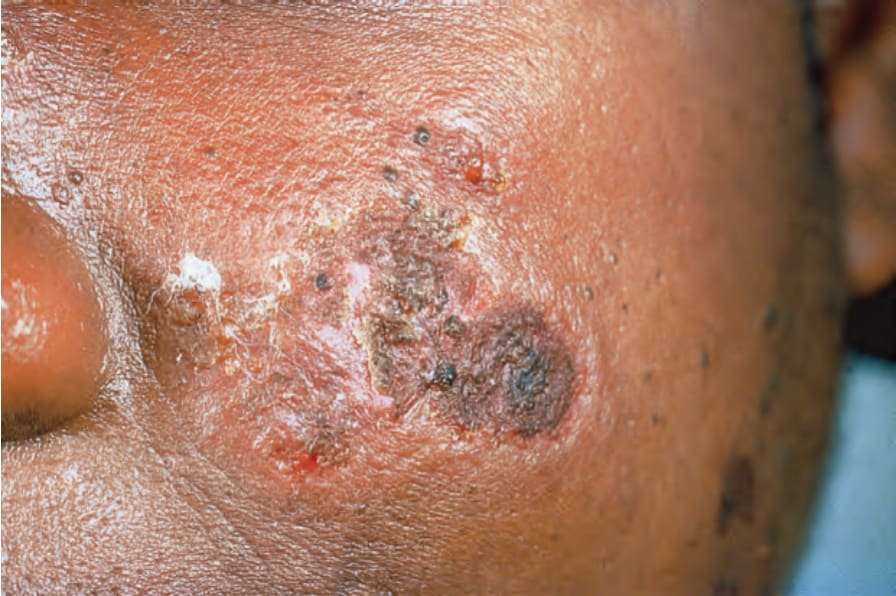
圖 17-12:盤狀紅斑性狼瘡——特寫。
Fig. 17.12 Discoid lupus erythematosus: close-up view. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
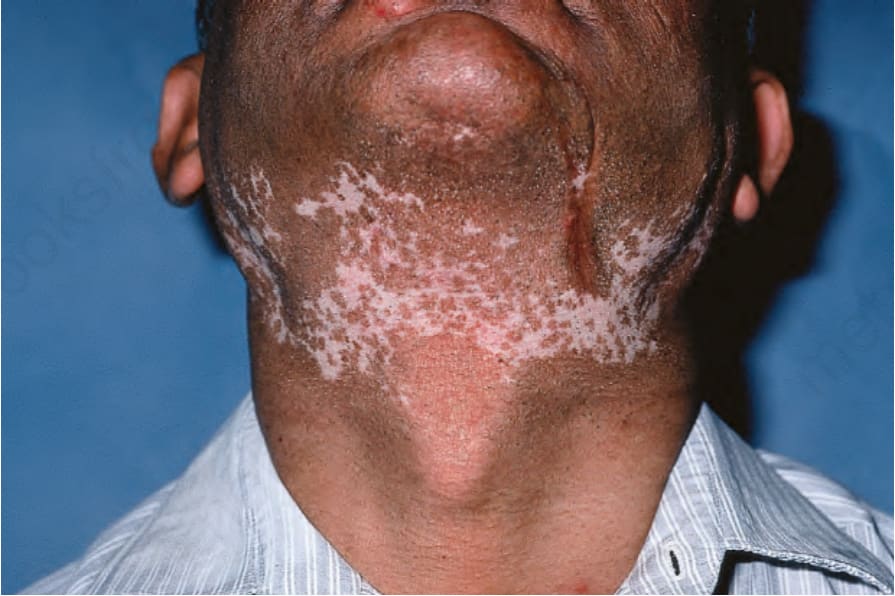
圖 17-13:盤狀紅斑性狼瘡——明顯的色素減少可能是令人困擾的併發症。
Fig. 17.13 Discoid lupus erythematosus: marked hypopigmentation may be a distressing complication. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
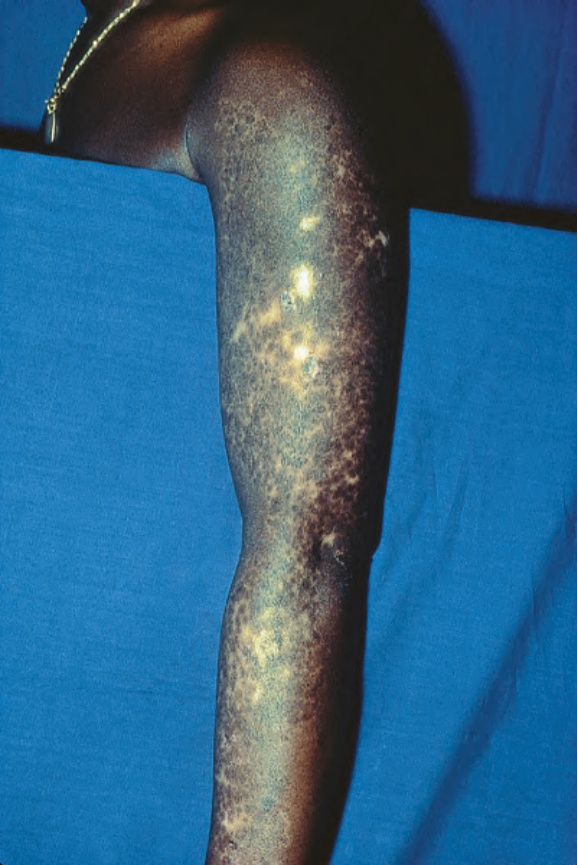
圖 17-14:盤狀紅斑性狼瘡——此病人的手臂受嚴重侵犯。
Fig. 17.14 Discoid lupus erythematosus: in this patient, the arm is severely affected. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
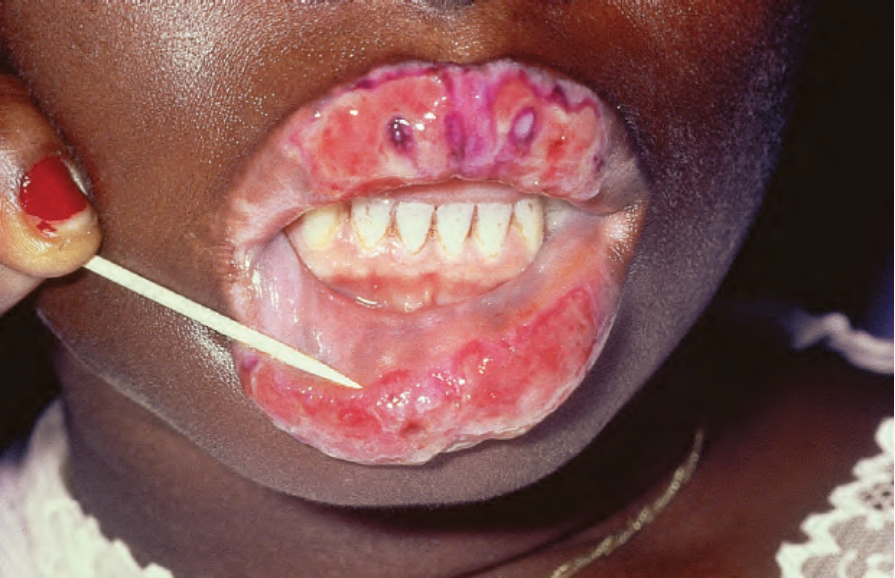
圖 17-15:盤狀紅斑性狼瘡——有嚴重的潰瘍性唇炎。上唇已用龍膽紫 (gentian violet) 染色。
Fig. 17.15 Discoid lupus erythematosus: there is gross ulcerative cheilitis. The upper lip has been stained with gentian violet. By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
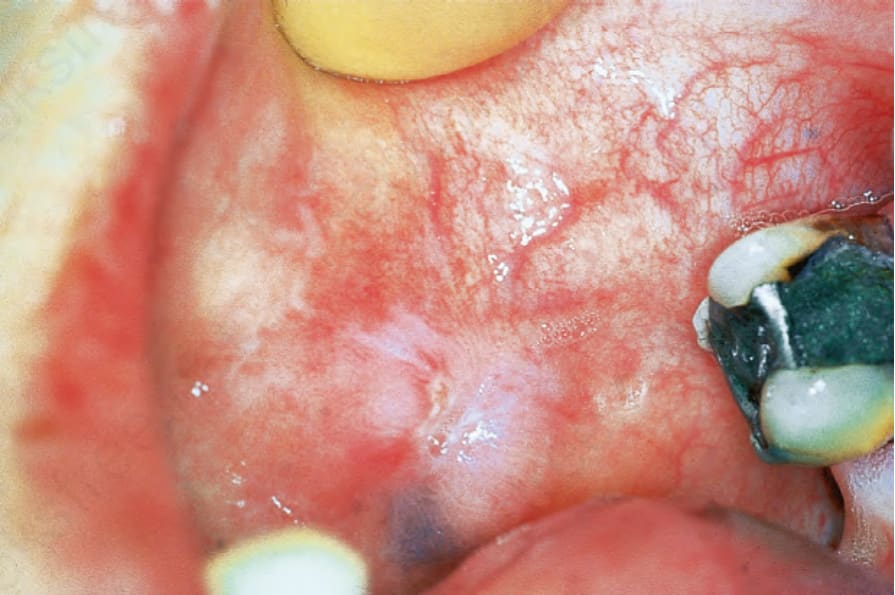
圖 17-16:盤狀紅斑性狼瘡——頰黏膜上的典型瘢痕性病灶。
Fig. 17.16 Discoid lupus erythematosus: typical scarred lesion on the buccal mucosa. By courtesy of R.A. Marsden, St George’s Hospital, London, UK.
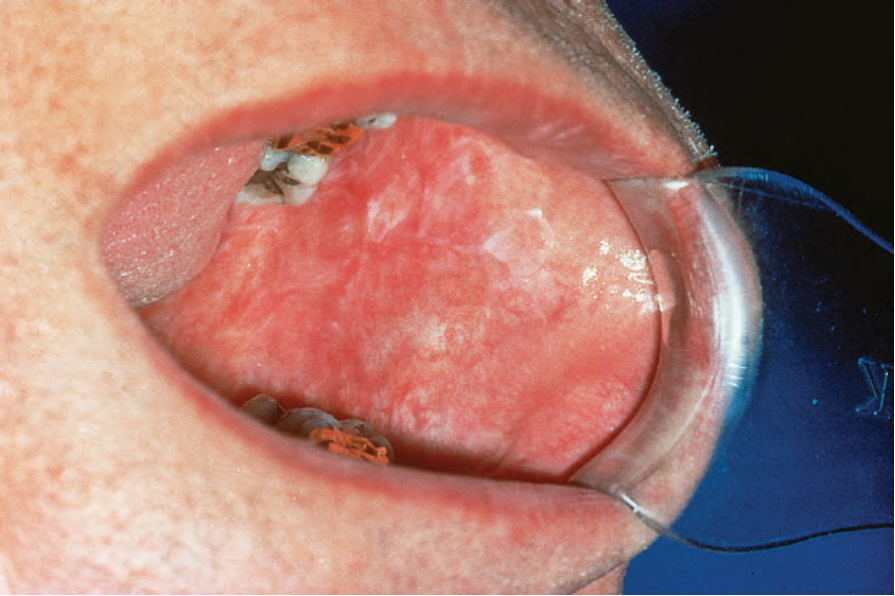
圖 17-17:盤狀紅斑性狼瘡——口腔侵犯有時難以與扁平苔癬 (lichen planus) 區分。
Fig. 17.17 Discoid lupus erythematosus: oral involvement is sometimes difficult to distinguish from lichen planus. By courtesy of the Institute of Dermatology, London, UK.
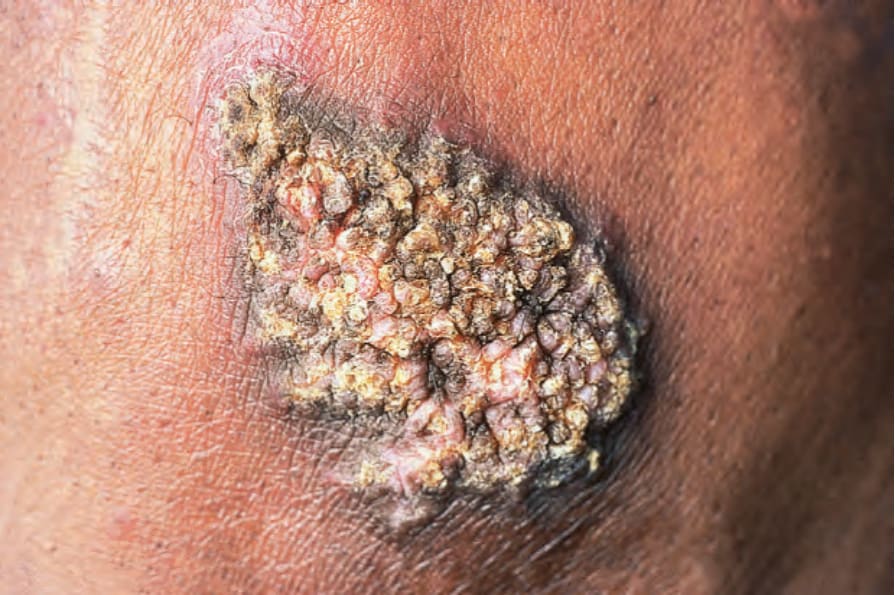
圖 17-18:疣狀盤狀紅斑性狼瘡——此病灶具有非常明顯的疣狀外觀。
Fig. 17.18 Verrucous discoid lupus erythematosus: this lesion has a very warty appearance. By courtesy of the Institute of Dermatology, London, UK.
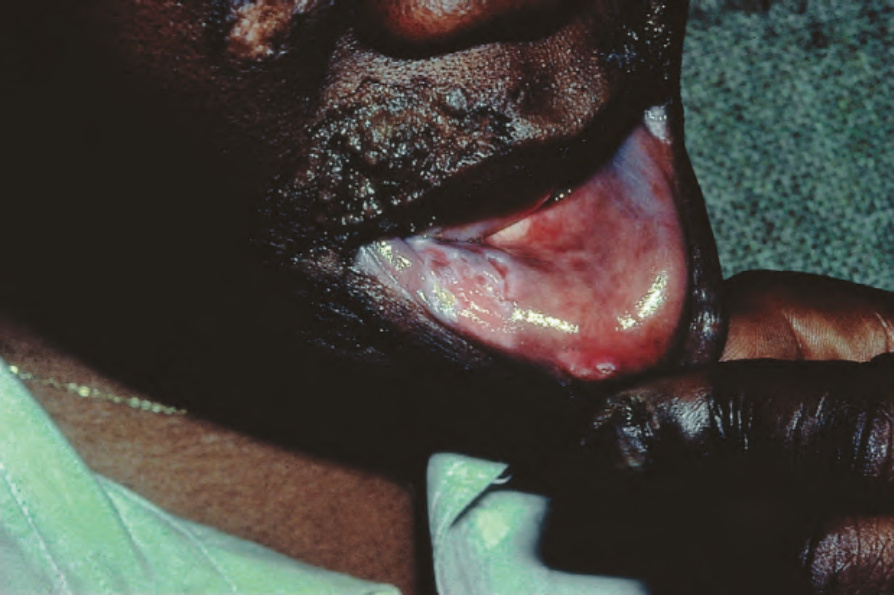
圖 17-19:疣狀盤狀紅斑性狼瘡——廣泛而毀容的疣狀斑塊,侵犯上唇與口角。
Fig. 17.19 Verrucous discoid lupus erythematosus: extensive disfiguring warty plaque involving the upper lip and angle of mouth. By courtesy of J. Newton Bishop, MD, St James’s University Hospital, Leeds, UK.
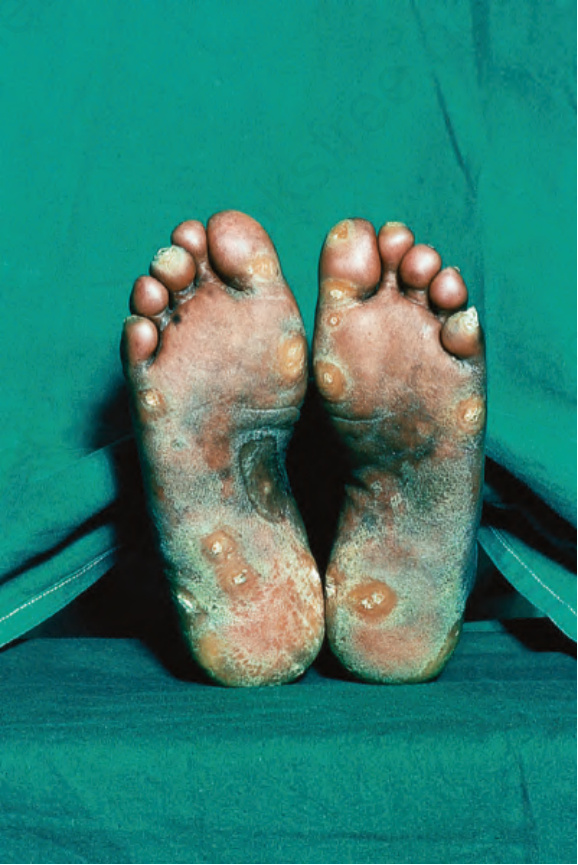
圖 17-20:疣狀盤狀紅斑性狼瘡——注意嚴重的過度角化 (hyperkeratosis)。
Fig. 17.20 Verrucous discoid lupus erythematosus: note the gross hyperkeratosis. By courtesy of J. Newton Bishop, MD, St James’s University Hospital, Leeds, UK.
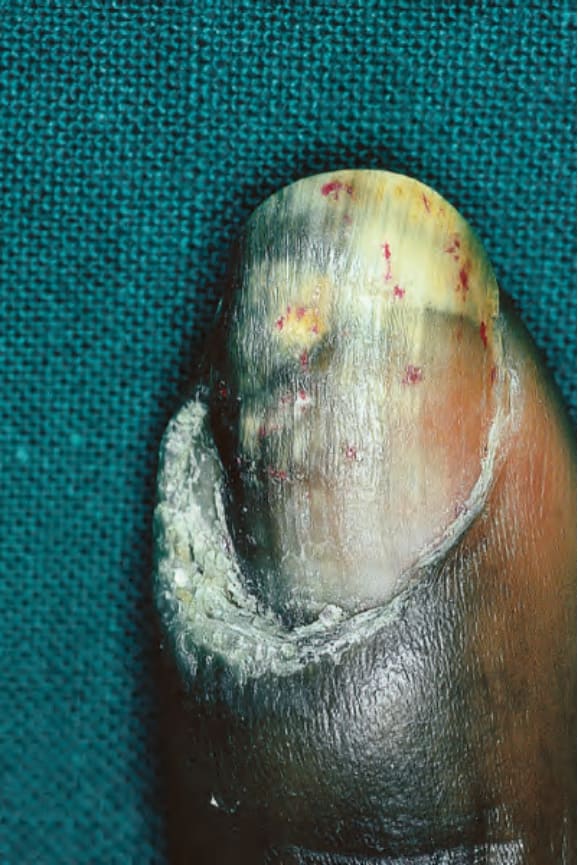
圖 17-21:疣狀盤狀紅斑性狼瘡——注意肥厚的甲上皮 (cuticle)、凹陷 (pitting) 與甲剝離 (onycholysis)。
Fig. 17.21 Verrucous discoid lupus erythematosus: note the hypertrophic cuticle, pitting, and onycholysis. By courtesy of J. Newton Bishop, MD, St James’s University Hospital, Leeds, UK.
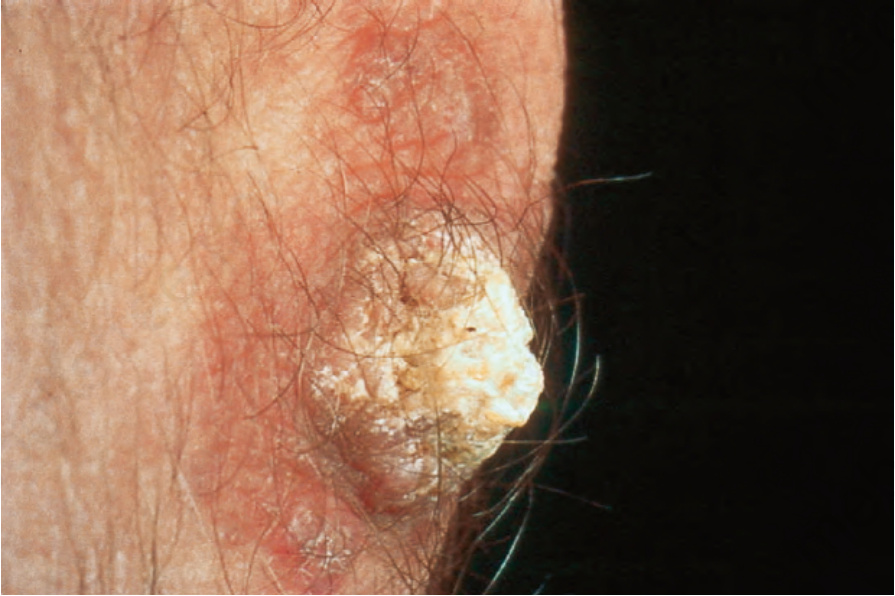
圖 17-22:疣狀盤狀紅斑性狼瘡——此例類似角化棘皮瘤 (keratoacanthoma)。
Fig. 17.22 Verrucous discoid lupus erythematosus: this example resembles a keratoacanthoma. By courtesy of the Institute of Dermatology, London, UK.
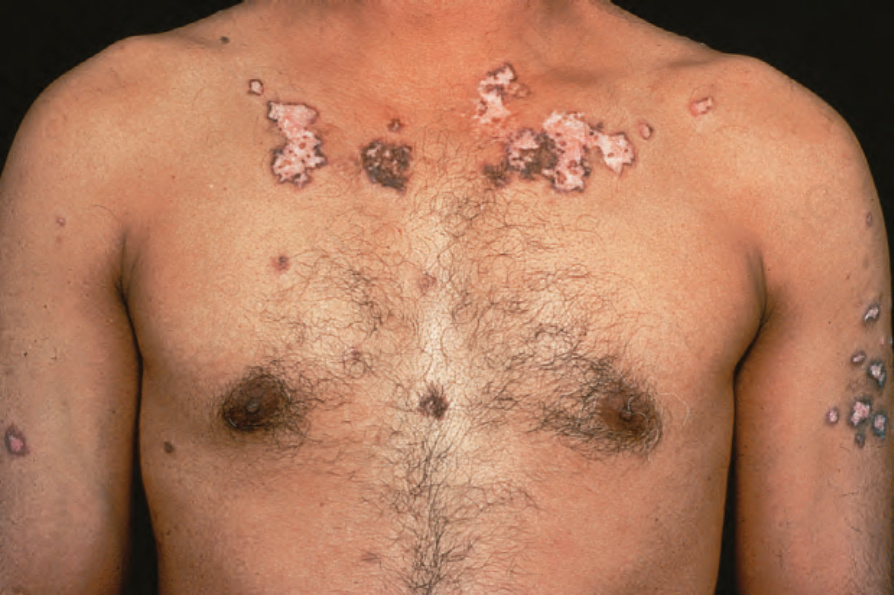
圖 17-23:泛發型盤狀紅斑性狼瘡——在此變異型中,病灶廣泛,可能侵犯胸部、肩部與上肢。注意廣泛的紅斑與鱗屑。
Fig. 17.23 Generalized discoid lupus erythematosus: in this variant, lesions are widespread and may involve the chest, shoulders, and upper limbs. Note the extensive erythema and scaling. By courtesy of R.A. Marsden, St George’s Hospital, London, UK.
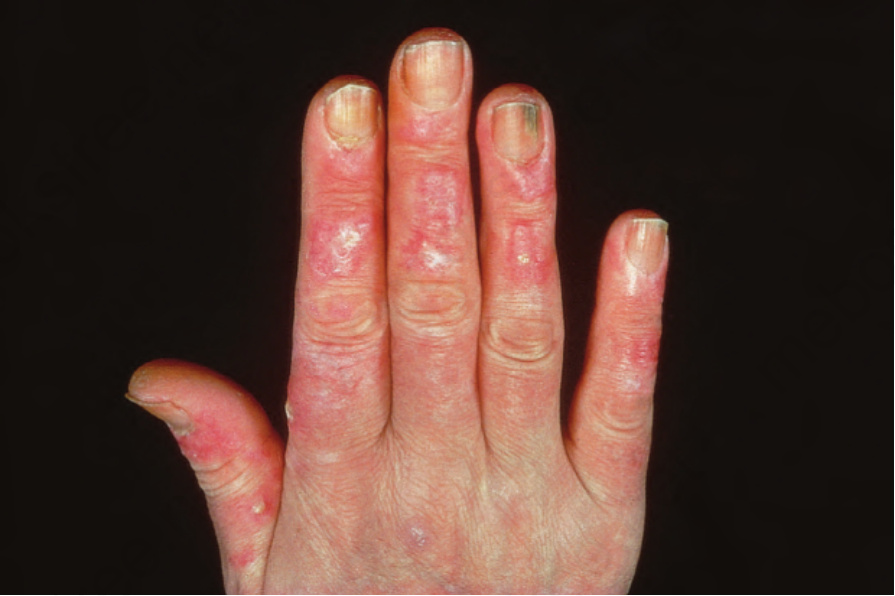
圖 17-24:泛發型盤狀紅斑性狼瘡——注意廣泛的紅斑與鱗屑。
Fig. 17.24 Generalized discoid lupus erythematosus: note the extensive erythema and scaling. By courtesy of R.A. Marsden, St George’s Hospital, London, UK.
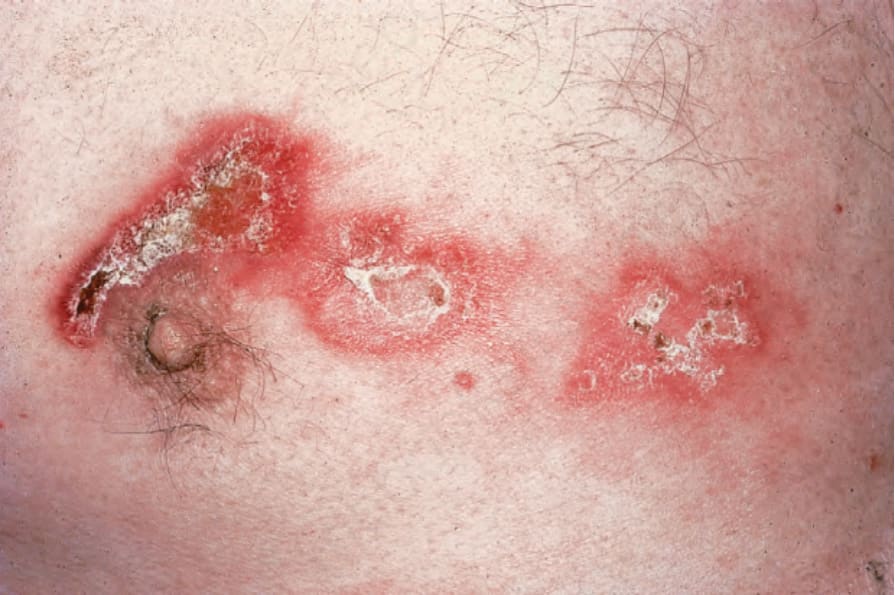
圖 17-25:泛發型盤狀紅斑性狼瘡——此病人胸部受嚴重侵犯。
Fig. 17.25 Generalized discoid lupus erythematosus: in this patient, the chest is severely affected. By courtesy of the Institute of Dermatology, London, UK.
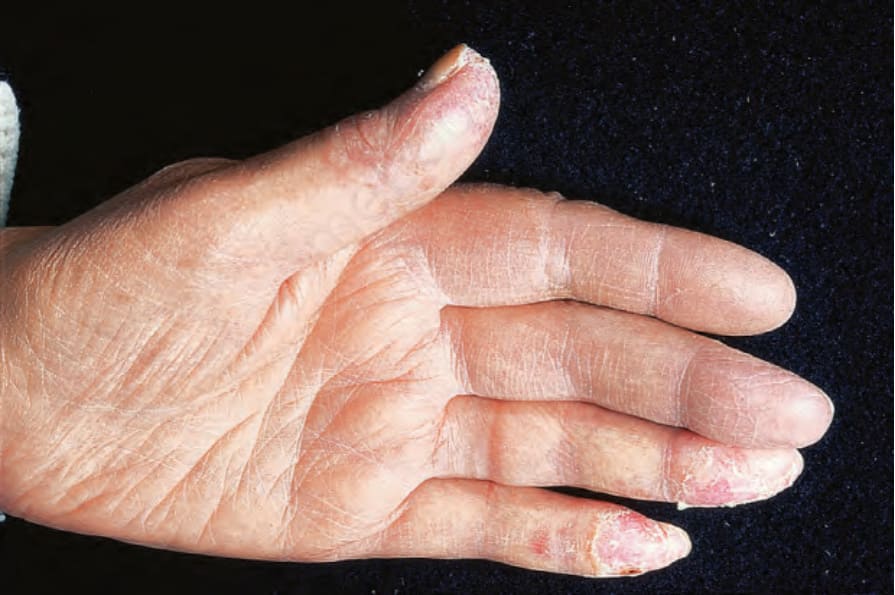
圖 17-26:凍瘡型紅斑性狼瘡——正在消退的凍瘡 (perniosis),侵犯拇指與無名指、小指的指尖。
Fig. 17.26 Chilblain lupus erythematosus: resolving perniosis involving the tips of the thumb and ring and little fingers. By courtesy of R.A. Marsden, St George’s Hospital, London, UK.
紅斑性狼瘡-多形性紅斑樣症候群 (Lupus erythematosus-erythema multiforme-like syndrome)
紅斑性狼瘡-多形性紅斑樣症候群(Rowell 症候群,Rowell syndrome)罕見。病人多為中年女性,除了具有任何變異型紅斑性狼瘡的特徵外,還在四肢,以及較少見的臉部、頸部、胸部與口腔,反覆發生環狀病灶。手掌與足底受侵犯屬例外。早期病灶為紅斑性丘疹,會變成環狀,並可能在邊緣形成水疱。此病通常癒合而不留瘢痕,但在嚴重疾病中可能發展出大疱,繼而壞死並潰瘍。此變異型的病人也有紅紺症 (erythrocyanosis)、凍瘡與 Raynaud 現象。有人提出 Rowell 症候群與 SLE 合併毒性表皮壞死溶解 (toxic epidermal necrolysis) 樣皮膚病灶代表同一疾病譜的不同兩端,後者多數案例是由藥物所誘發。紅斑性狼瘡-多形性紅斑樣症候群病人的血清含有斑點型抗核因子(最一致的特徵,存在於 88% 的病人)、類風濕因子,以及 anti-Ro/La 抗體。在罕見案例中,anti-Ro/La 與類風濕因子為陰性。與抗磷脂症候群 (antiphospholipid syndrome) 的關聯屬例外性記載。
深部紅斑性狼瘡 (Lupus erythematosus profundus)
深部紅斑性狼瘡 (lupus erythematosus profundus,脂膜炎 panniculitis) 是另一個不常見的變異型,可能與 DLE 或 SLE 相關而發生。此於第 10 章有詳細討論。
DLE 與慢性肉芽腫病 (DLE and chronic granulomatous disease)
偶爾,DLE 樣皮膚病會發生於 X 染色體連鎖慢性肉芽腫病 (X-linked chronic granulomatous disease) 的女性帶因者。受影響兒童的母親偶爾可顯示類似病灶,表現為臉部與手部藍紅色、浸潤性、有鱗屑的丘疹,有時伴隨光敏性。額外特徵包括復發性阿弗他樣 (aphthous-like) 潰瘍性口炎與凍瘡。
X 染色體連鎖慢性肉芽腫病以隱性性狀遺傳,病人(通常為男孩)因嗜中性球殺菌活性缺陷而有嚴重且反覆的感染。異型合子的女性帶因者顯示部分缺陷,可由 nitroblue tetrazolium 還原減少所指示,但通常無感染。亦有記載一種可能出現盤狀狼瘡樣病灶的自體隱性變異型。DLE 病灶在帶因者與慢性肉芽腫病病人皆有報告;然而,慢性肉芽腫病病人發展為 SLE 極為罕見。亞急性皮膚紅斑性狼瘡樣病灶與腫脹性狼瘡 (lupus tumidus) 也可能發生。DLE 亦曾被描述於非 X 染色體連鎖的高 IgM 症候群 (hyper-IgM syndrome)。
亞急性皮膚紅斑性狼瘡 (Subacute cutaneous lupus erythematosus)
亞急性皮膚紅斑性狼瘡 (subacute cutaneous lupus erythematosus, SCLE) 占紅斑性狼瘡病人的 5% 至 10%。約有 50% 依據修訂版美國風濕學會 (American Rheumatology Association) 診斷標準(見下文)符合 SLE。SCLE 在白種人中占多數,在黑人、韓國人與中國人中不常見。女性受影響的頻率高於男性(2.3 : 1),表現的平均年齡約為 40 歲。此狼瘡變異型在兒童中非常罕見。曾有一名 SCLE 兒童被報告出現侵犯多數指甲的甲營養不良 (nail dystrophy)。
此皮疹常廣泛分布,由對稱性、非瘢痕性且非硬結性的紅斑鱗屑性病灶構成 (Fig. 17.27)。沿著 Blaschko 線的獨特表現也曾被報告。
無硬結 (induration) 被視為 SCLE 與 DLE 之間一個有用的臨床鑑別特徵。此外,缺乏毛囊栓塞、黏附性鱗屑與真皮萎縮,有助於將亞急性病灶與盤狀變異型區分。搔癢非常罕見。病灶可能變成丘疹鱗屑性(乾癬樣 psoriasiform)或環狀,後者融合而產生多環狀 (polycyclic) 與迴旋狀 (gyrate) 的形態 (Fig. 17.28)。在部分病人,兩種型態皆可見。在環狀病灶的活動性邊緣,有時可見結痂與水疱。也曾描述糠疹樣 (pityriasiform) 與多形性紅斑樣病灶,以及偶發的慢性白斑 (leukoderma)。例外情況下,此病可能表現為泛發性皮膚異色症 (poikiloderma) 或紅皮症 (erythroderma)。此外,15% 至 30% 的病人會發展出典型 DLE 的特徵,通常位於頭皮或臉部。有時也可見 SLE 的顴部紅斑 (malar erythema)(15%)。輕微色素減少、毛細血管擴張、非瘢痕性禿髮、網狀青斑 (livedo reticularis)、Raynaud 現象與黏膜潰瘍是額外的特徵。營養不良性鈣化屬例外。皮膚白血球碎裂性血管炎 (leukocytoclastic vasculitis) 見於少數病人,似乎為自限性,且不伴隨惡化的預後。
光敏性極為重要,因此病灶典型上見於臉部、頸部、上背與胸部、肩部、手臂伸側、手背與手指 (Figs 17.29 and 17.30)。
SCLE 病人有較高的人類白血球抗原 (human leukocyte antigen, HLA)-DR3(75%)、HLA-B8 與 HLA-A1 發生率,並與遺傳性同型合子 C2 與 C4 缺乏有顯著關聯。HLA-DR2 也以較高頻率出現,尤其見於丘疹鱗屑性而非環狀皮膚病灶者。抗核抗體 (antinuclear antibodies) 見於約 50% 的病人,anti-Ro (SS-A) 抗體則存在於約 65%,尤其是那些環狀多環病灶者。anti-La (SS-B) 抗體也常明顯可見。SCLE 兒童多數案例通常顯示 anti-Ro (SS-A) 抗體,anti-La (SS-B) 抗體則少見。此外,超過 70% 的兒童 SCLE 病人可偵測到 ANA。
SCLE 的病程傾向於相對良性,但全身性表現相當常見且可能嚴重。嚴重的皮膚外疾病似乎較常見於罹患丘疹鱗屑性 SCLE 的男性。然而,皮膚病灶的類型並非總是被證實與皮膚外表現的嚴重度相關。腎臟疾病曾被報告為不常見,但近期一項研究發現其頻率與 SLE 相似且同樣嚴重。罕見情況下,病人會發展出 SLE 的其他更嚴重表現。
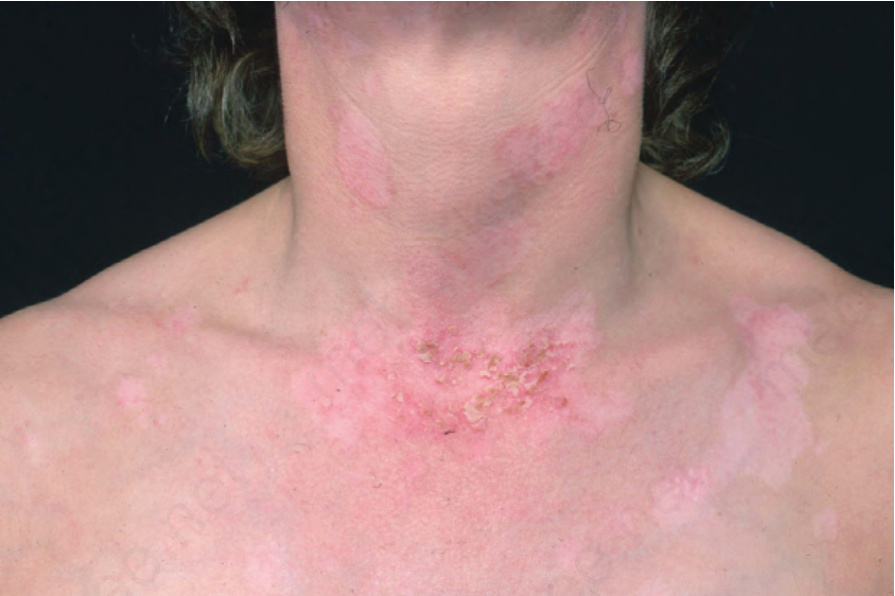
圖 17-27:亞急性皮膚紅斑性狼瘡——環狀病灶,許多已癒合並伴有色素變化,活動性病灶上覆有細緻的鱗屑。
Fig. 17.27 Subacute cutaneous lupus erythematosus: annular lesions, many healed with pigmentary changes and delicate scale overlying an active lesion. From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
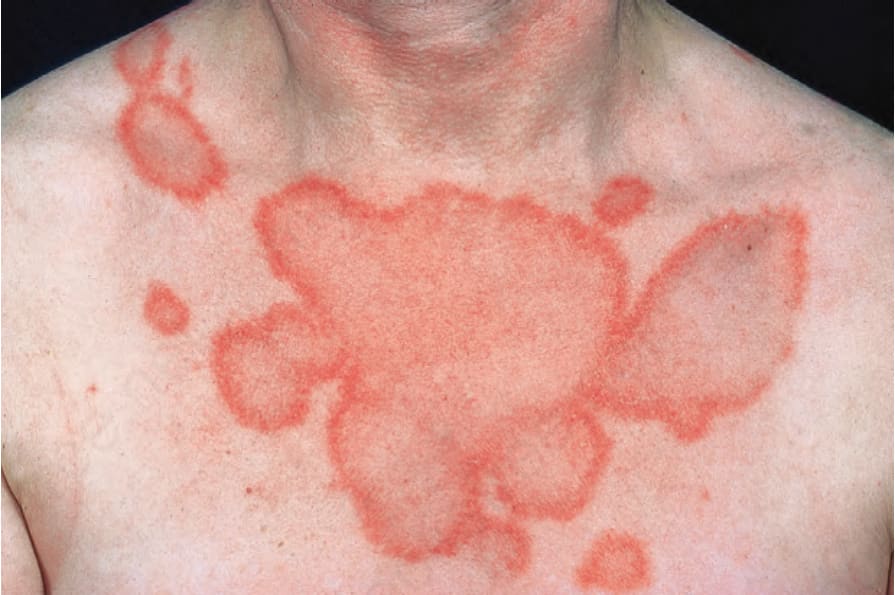
圖 17-28:亞急性皮膚紅斑性狼瘡——環狀病灶融合造成此奇特的皮疹。
Fig. 17.28 Subacute cutaneous lupus erythematosus: coalescence of annular lesions has resulted in this bizarre eruption. By courtesy of the Institute of Dermatology, London, UK.
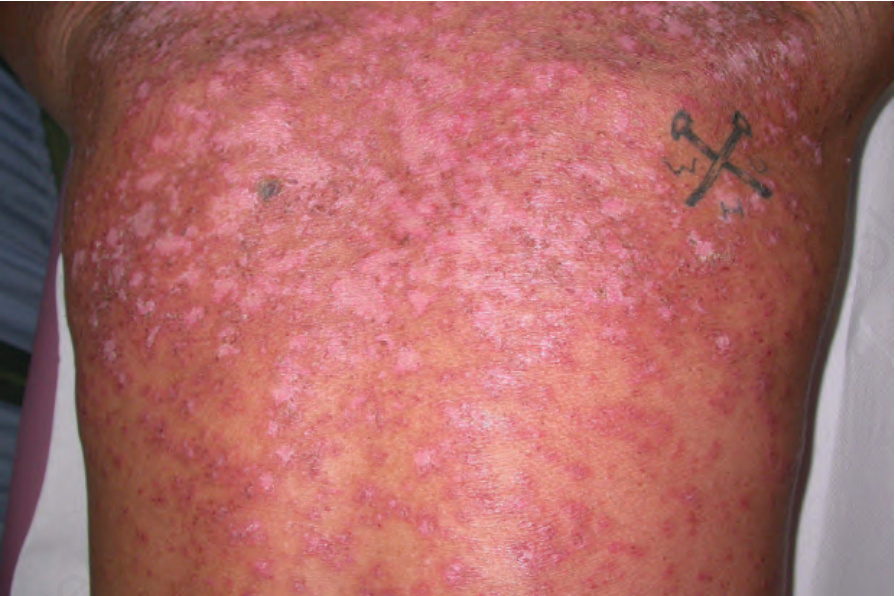
圖 17-29:亞急性皮膚紅斑性狼瘡——此病人有非常廣泛的侵犯。
Fig. 17.29 Subacute cutaneous lupus erythematosus: in this patient, there is very extensive involvement. By courtesy of Dr J.C. Pascual, Alicante, Spain.
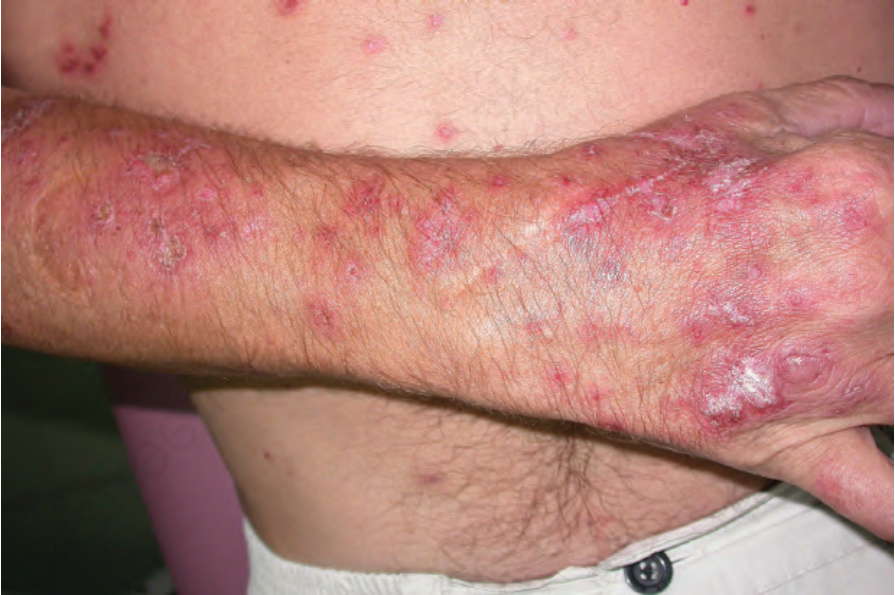
圖 17-30:亞急性皮膚紅斑性狼瘡——病灶呈強烈紅斑,並有明顯鱗屑。
Fig. 17.30 Subacute cutaneous lupus erythematosus: the lesions are intensely erythematous and there is marked scaling. By courtesy of Dr J.C. Pascual, Alicante, Spain.
SCLE 病人有較高的類風濕性關節炎 (rheumatoid arthritis) 與 Sjögren 症候群 (Sjögren syndrome) 發生率。合併 SCLE 與 Sjögren 症候群者具有高效價 anti-Ro 抗體、高皮膚血管炎發生率,以及嚴重神經精神與肺部侵犯的風險增加。SCLE 曾被記載與下列情況相關:遲發性皮膚紫質症 (porphyria cutanea tarda)、B 型肝炎病毒感染、放射治療、放射碘 (radioiodine) 治療、頭頸部鱗狀細胞癌,以及乳房、肝細胞與肺臟之癌瘤。與包涵體肌炎 (inclusion body myositis) 及伴有粒線體變化的間質性肌炎 (interstitial myositis) 之例外性關聯亦有記載。與扁平苔癬及菊池-藤本病 (Kikuchi-Fujimoto disease) 的關係屬例外。臨床與組織學上完全相同的案例曾記載於下列藥物治療之後:hydrochlorothiazide、griseofulvin、抗組織胺、terbinafine、鈣離子通道阻斷劑、nifedipine、angiotensin-converting enzyme (ACE) 抑制劑、質子幫浦抑制劑、interferon、phenytoin、bupropion、ticlopidine、capecitabine、lansoprazole、leflunomide、fluorouracil、leuprorelin、efalizumab、acebutolol、tamoxifen、docetaxel、statin、citalopram、golimumab、rituximab、gemcitabine、adalimumab、bevacizumab、paclitaxel、ranibizumab、terbinafine、pazopanib、norfloxacin、minocycline、pemetrexed 加 carboplatin 化療、imiquimod、mitotane,以及 anastrozole。
全身性紅斑性狼瘡的嗜中性球皮膚病 (Neutrophilic dermatosis of the systemic lupus erythematosus)
SLE 病人,以及 hydralazine 誘發性狼瘡與新生兒狼瘡病人,偶爾會發展出一種罕見的皮膚皮疹,形態學上模擬 Sweet 症候群 (Sweet syndrome)。文獻中對此情況使用過數個替代名稱,例如嗜中性球皮膚病 (neutrophilic dermatosis) 或嗜中性球蕁麻疹性皮膚病 (neutrophilic urticarial dermatosis),後者源於其與蕁麻疹的臨床相似性。嗜中性球皮膚病在 SLE 病人中的頻率估計低於 5%。然而重要的是,嗜中性球皮膚病可在多達三分之一的病人中作為 SLE 的初始表現。儘管如此,此反應型態並非 SLE 所特有,也可見於多種不同疾病,例如類風濕性關節炎、Sjögren 症候群、cryopyrin 相關週期性症候群 (cryopyrin-associated periodic syndromes)、Schnitzler 症候群與成人發作型史迪爾病 (adult-onset Still disease)。
病人表現為紅斑性、粉紅色或紫紅色不癢的斑塊,或略為隆起的丘疹。無黏膜侵犯。好發部位包括四肢,其次為軀幹、臉部以及頭頸部區域。掌蹠侵犯亦有報告。所有年齡層皆可受影響,從新生兒期到老年病人皆有,但此皮疹最常發展於生命的第四個十年。SLE 與新生兒狼瘡病人有顯著的女性優勢,而 hydralazine 誘發性狼瘡則典型上發生於男性。包括發燒、倦怠與關節炎在內的全身性症狀,存在於部分病人。
全身性紅斑性狼瘡 (Systemic lupus erythematosus)
除了多變的皮膚表現外,SLE 的病灶幾乎可在身體的任何器官或系統發現。美國風濕學會的指引在建立診斷上很有價值:任何曾經歷四項或以上標準(無論是先後或同時)的病人,皆被視為患有 SLE (Table 17.2)。儘管如此,2012 年提出了系統性狼瘡國際合作臨床中心 (Systemic Lupus International Collaborating Clinics, SLICC) 之 SLE 分類標準,代表對先前 ARA 指引的演進與精煉。SLICC 標準將標準總數從 11 項增加到 17 項 (Table 17.3)。要被診斷為 SLE,需符合下列標準:(1) 符合至少四項標準(其中至少一項臨床與一項免疫學標準),或 (2) 在 ANA 或 anti-dsDNA 抗體存在下,以狼瘡腎炎 (lupus nephritis) 作為唯一標準。
SLE 的特徵為顯著的女性優勢(9 : 1),通常於第三、第四與第五個十年表現。在非裔加勒比人 (Afro-Caribbeans) 中發生率高,牙買加女性的最高盛行率為 1/150。在美國,發生率約為 1/100。
皮膚侵犯發生於 75% 至 88% 的病人。病灶高度多形,可能模擬許多其他皮膚病 (Table 17.4)。「蝴蝶斑」(butterfly rash) 為典型表現,是一種略有鱗屑、有時呈水腫性的紅斑,特別分布於鼻樑與臉頰 (Fig. 17.31)。光敏性常見,影響超過 50% 的病人,紅斑性或紫紅色斑丘疹性皮疹可能發展,尤其在白種病人,在其他光暴露區域,如頸部的「V」形區域與前臂。對紫外線 (ultraviolet, UV) A 與 B 光皆敏感。約 15% 的病人,特別是非裔加勒比人,有類似 DLE 的病灶,這顯然與較不嚴重的疾病及較低的腎臟侵犯頻率相關。SCLE 樣特徵存在於 10% 至 15% 的病人。有人提出,合併 SLE 與 SCLE 病灶的病人具有較有利的預後。
禿髮很重要,發生於約 20% 的病人,可能為瘢痕性或非瘢痕性。較常見的非瘢痕性病灶,往往構成相當瀰漫的掉髮,作為對壓力的非特異性反應而發生(休止期落髮 telogen effluvium)。額部頭髮斷裂 (fractured frontal hairs) 是特徵性表現。
Raynaud 現象發生於 10% 至 40% 的病人。紫斑與瘀斑常見,部分可能源於 corticosteroid 治療,但血小板低下與免疫複合體媒介的血管炎在致病機轉中也扮演角色。血管炎發生於多達 30% 的病人,也可能造成梗塞、潰瘍、指部結節、瘢痕與壞疽 (Figs 17.32–17.34)。特別影響手臂與大腿以及關節周圍部位的網狀青斑,可能是多達 10% 病人的表現特徵,而白色萎縮 (atrophie blanche) 的變化偶有記載(livedoid vasculitis)(Fig. 17.35)。網狀青斑常是抗心磷脂症候群 (anticardiolipin syndrome) 的特徵,偶爾也有人描述與 Degos 病(惡性萎縮性丘疹病 malignant atrophic papulosis)所見相同的表現病灶。血管炎特徵與腎臟及中樞神經系統侵犯相關。蕁麻疹常被發現。
紅斑性肢痛症 (erythromelalgia),一種暴露於熱之後伴隨紅斑的灼熱感,也曾被注意到。侷限性與瀰漫性色素增加以及蕁麻疹性病灶(包括蕁麻疹性血管炎 urticarial vasculitis)較不常見。除了梗塞與潰瘍外,指部表現包括甲周與指關節紅斑、甲褶毛細血管擴張 (nail fold telangiectases)、甲上皮病灶與裂片狀出血 (splinter hemorrhages)。毛細血管擴張也可見於指尖與手掌。部分病人發展出紅色甲弧 (red lunulae)。上述相關的多形性紅斑樣與凍瘡樣病灶已於前文描述。黏膜侵犯發生於約 10% 的病人:無痛性潰瘍最常見,但其他特徵包括紅斑、瘀點、糜爛與出血。硬腭中央、唇部與頰黏膜特別受侵犯。
偶爾可見的額外病灶包括持續性眼瞼水腫與紅斑、類風濕樣結節、真皮黏液沉著 (dermal mucinosis)、大疱型變異型、血栓性血小板低下性紫斑 (thrombotic thrombocytopenic purpura)、混合型冷球蛋白血症 (mixed cryoglobulinemia),以及軟組織鈣化。SLE 中的水疱與大疱可能併發於極度的基底細胞水樣變性、代表自體免疫大疱性皮膚病的共同表現,或代表一種特定的疱疹樣皮膚炎樣皮疹(SLE 的大疱性皮膚病 bullous dermatosis of SLE)。與遲發性皮膚紫質症有已知關聯。
約 5% 至 10% 的 SLE 病人抗核抗體陰性。這似乎與一個特定亞型相關。病人通常 HLA-DR3 陽性,並有 Sjögren 症候群,以及肺部侵犯、精神表現、皮膚血管炎與高γ球蛋白血症性紫斑 (hypergammaglobulinemic purpura) 之發生率增加。
非特異性症狀常見,包括慢性疲倦、體重減輕、發燒、倦怠與虛弱。關節痛與肌痛發生於 90% 的病人。肌痛可能造成失能,但客觀的肌肉無力罕見。同樣地,雖然關節痛可能明顯,但通常少有關節損傷的臨床證據。關節積液常見。約 25% 的病人有明顯的關節炎,可能為遊走性多關節炎或慢性進行性多關節炎並伴有變形。病人有時因原發疾病或類固醇治療而發展出缺血性骨壞死 (avascular bone necrosis)。極罕見情況下,肌腱侵犯伴隨攣縮的發展。
心血管系統的病灶表現為心臟肥大、心包炎、心包積液與/或心內膜炎(Libman-Sacks 瓣膜炎,Libman-Sacks valvulitis)。在 SLE 中,二尖瓣主要受侵犯,但具有狼瘡抗凝血劑症候群 (lupus anticoagulant syndrome) 的病人似乎有發展為主動脈瓣疾病的特別風險。傳導缺陷與充血性心衰竭是額外的特徵。
呼吸系統侵犯可能表現為胸膜炎(伴或不伴積液)、細菌性肺炎,或極罕見的狼瘡性肺炎 (lupus pneumonitis)。肺高壓 (pulmonary hypertension) 估計存在於 0.5% 至 14% 的 SLE 病人,尤其是那些伴有抗磷脂抗體者。
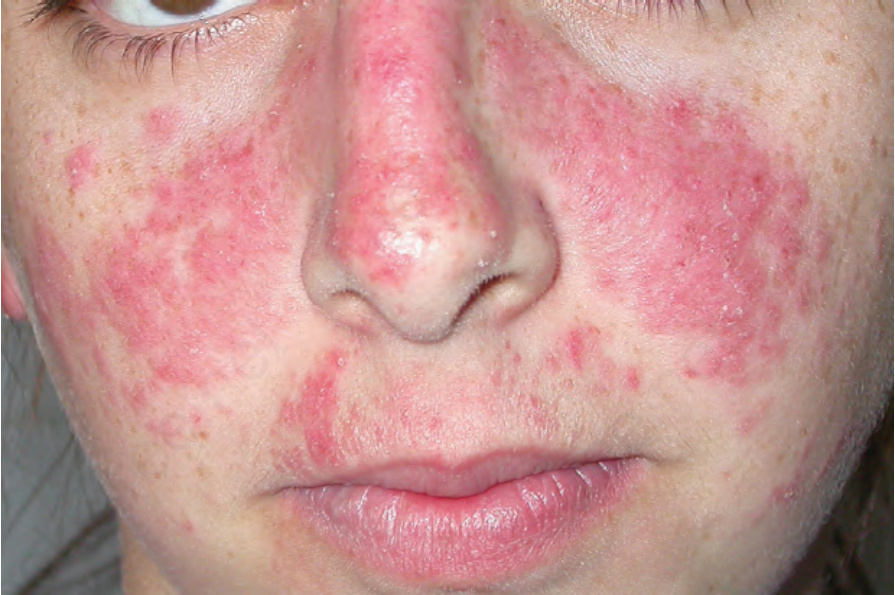
圖 17-31:全身性紅斑性狼瘡——臉頰與鼻部上特徵性的「蝴蝶紅斑」(butterfly erythema)。
Fig. 17.31 Systemic lupus erythematosus: characteristic ‘butterfly erythema’ on the cheeks and nose. By courtesy of Dr J.C. Pascual, Alicante, Spain.
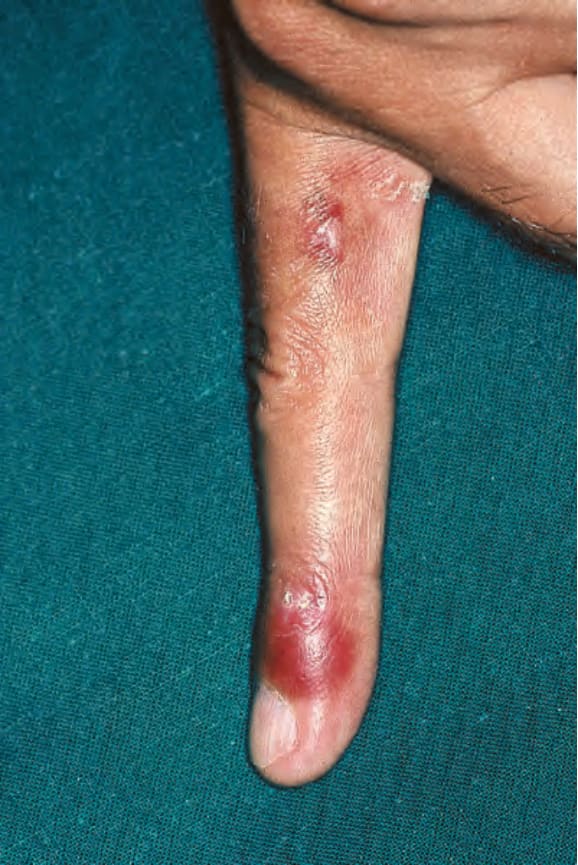
圖 17-32:全身性紅斑性狼瘡——指尖上的紅斑性血管炎病灶。
Fig. 17.32 Systemic lupus erythematosus: erythematous vasculitic lesion on the fingertip. By courtesy of M.M. Black, MD, St Thomas’ Hospital, London, UK.
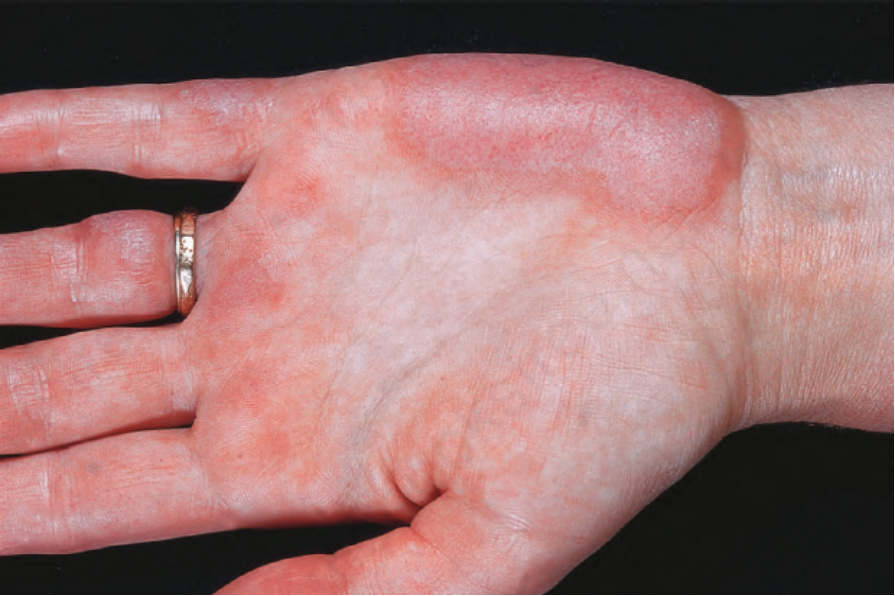
圖 17-33:全身性紅斑性狼瘡——手掌侵犯罕見,可能為血管炎性。
Fig. 17.33 Systemic lupus erythematosus: involvement of the palms is rare and may be vasculitic. By courtesy of the Institute of Dermatology, London, UK.
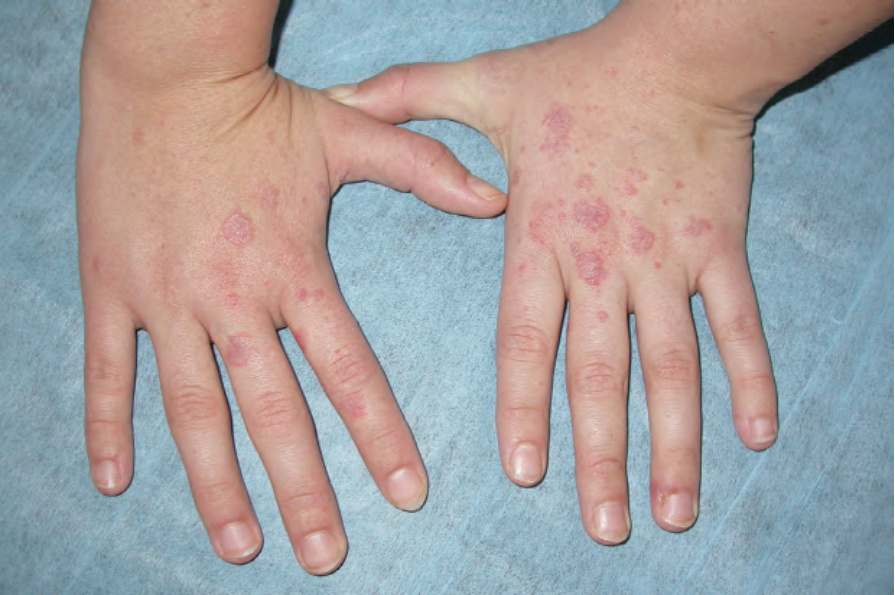
圖 17-34:全身性紅斑性狼瘡——手背與手指上有紅斑性結節。
Fig. 17.34 Systemic lupus erythematosus: erythematous nodules are present on the back of the hand and on the fingers. By courtesy of Dr J.C. Pascual, Alicante, Spain.
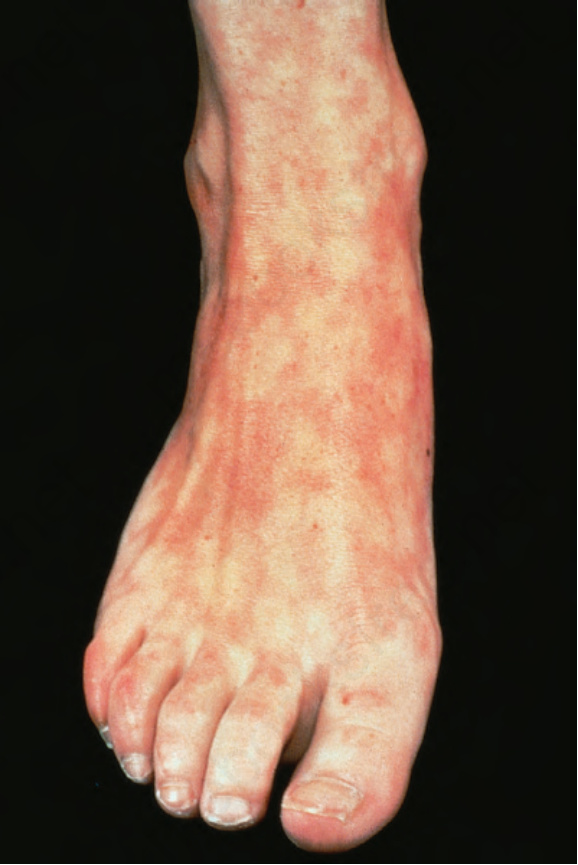
圖 17-35:全身性紅斑性狼瘡——網狀青斑 (livedo reticularis) 因分水嶺區的相對缺血而發展。其成因眾多,特別是結締組織疾病。
Fig. 17.35 Systemic lupus erythematosus: livedo reticularis develops as a consequence of relative ischemia in the watershed zones. There are numerous causes, particularly connective tissue disease. By courtesy of the Institute of Dermatology, London, UK.
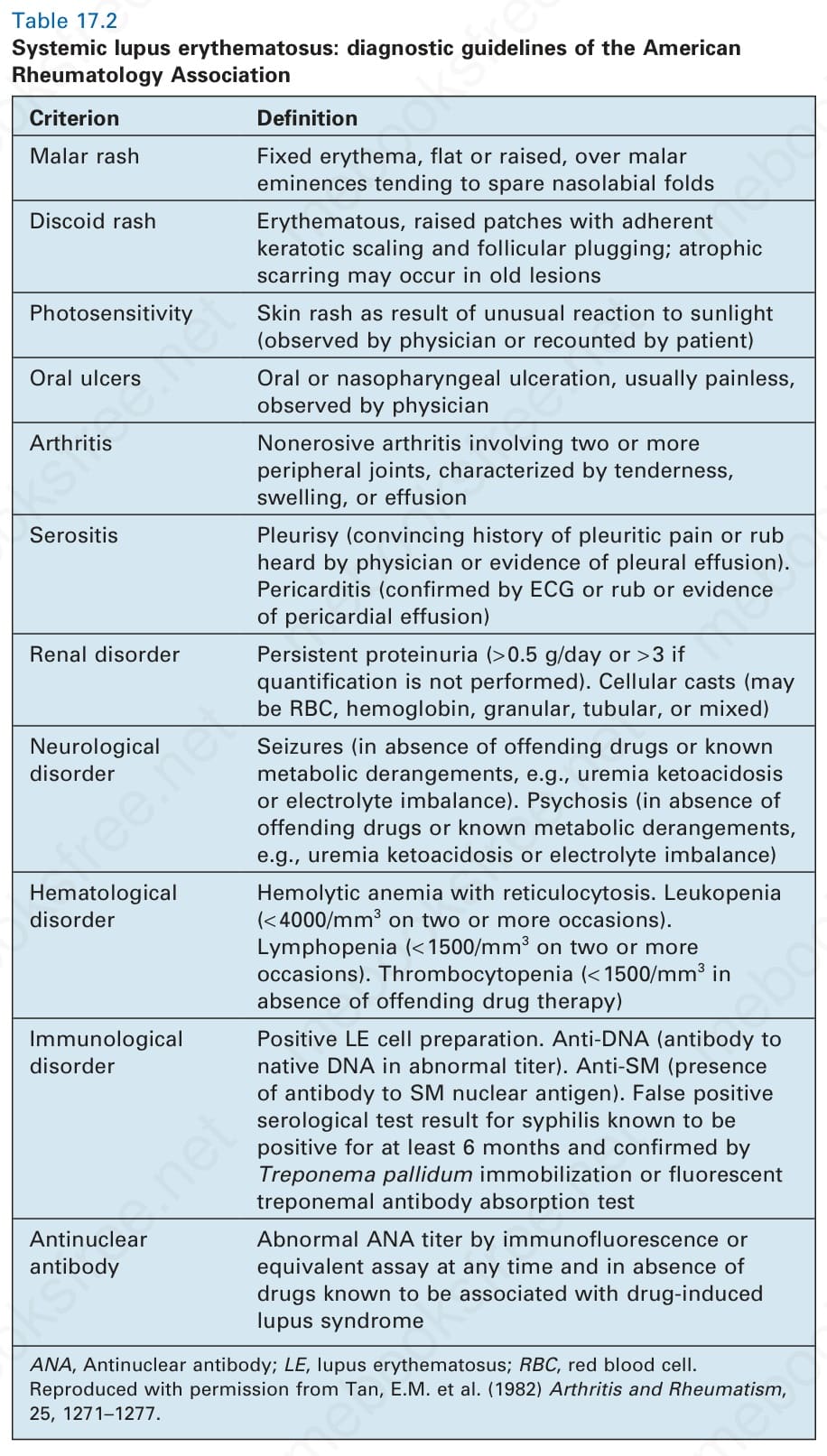
表 17-2:全身性紅斑性狼瘡——美國風濕學會的診斷指引 (diagnostic guidelines of the American Rheumatology Association)。
Table 17.2 Systemic lupus erythematosus: diagnostic guidelines of the American Rheumatology Association
下表列出美國風濕學會診斷標準(標準與定義):
- 顴部皮疹 (Malar rash):橫跨顴隆突的固定性紅斑,扁平或隆起,傾向不侵犯鼻唇溝。
- 盤狀皮疹 (Discoid rash):紅斑性、隆起的斑片,伴有黏附性角化鱗屑與毛囊栓塞;舊病灶可能發生萎縮性瘢痕。
- 光敏性 (Photosensitivity):因對日光的異常反應所致之皮膚皮疹(由醫師觀察到或由病人陳述)。
- 口腔潰瘍 (Oral ulcers):口腔或鼻咽潰瘍,通常無痛,由醫師觀察到。
- 關節炎 (Arthritis):非侵蝕性關節炎,侵犯兩個或以上的周邊關節,特徵為壓痛、腫脹或積液。
- 漿膜炎 (Serositis):胸膜炎(有令人信服的胸膜性疼痛病史,或由醫師聽到摩擦音,或有胸膜積液證據)。心包炎(由 ECG 或摩擦音確認,或有心包積液證據)。
- 腎臟疾患 (Renal disorder):持續性蛋白尿(> 0.5 g/day,若未進行定量則 > 3+)。細胞圓柱(可能為紅血球、血紅素、顆粒、管狀或混合型)。
- 神經疾患 (Neurological disorder):癲癇發作(在無致病藥物或已知代謝紊亂,如尿毒症酮酸中毒或電解質不平衡之情況下)。精神病(在無致病藥物或已知代謝紊亂,如尿毒症酮酸中毒或電解質不平衡之情況下)。
- 血液疾患 (Hematological disorder):溶血性貧血伴網狀紅血球增多。白血球低下(兩次或以上 < 4000/mm³)。淋巴球低下(兩次或以上 < 1500/mm³)。血小板低下(在無致病藥物治療之情況下 < 1500/mm³)。
- 免疫疾患 (Immunological disorder):LE 細胞製備陽性。Anti-DNA(對 native DNA 之抗體呈異常效價)。Anti-SM(存在對 SM 核抗原之抗體)。梅毒血清學檢驗偽陽性,已知陽性至少 6 個月,並經 Treponema pallidum 固定試驗或螢光梅毒螺旋體抗體吸收試驗確認。
- 抗核抗體 (Antinuclear antibody):免疫螢光或等效檢測法測得異常 ANA 效價,可於任何時間,且在無已知與藥物誘發性狼瘡症候群相關之藥物之情況下。
ANA,抗核抗體;LE,紅斑性狼瘡;RBC,紅血球。經許可重製自 Tan, E.M. et al. (1982) Arthritis and Rheumatism, 25, 1271–1277。

表 17-3:SLICC 分類標準所使用之臨床與免疫學標準 (clinical and immunological criteria used in the SLICC classification criteria)。
Table 17.3 Clinical and immunological criteria used in the SLICC classification criteria*
臨床標準 (Clinical criteria):
- 急性皮膚狼瘡 (Acute cutaneous lupus),包括狼瘡顴部皮疹(若顴部皮疹為盤狀則不計入);大疱型狼瘡 (bullous lupus);SLE 之毒性表皮壞死溶解變異型;斑丘疹性狼瘡皮疹;光敏性狼瘡皮疹(在無皮肌炎或亞急性皮膚狼瘡之情況下)。
- 慢性皮膚狼瘡 (Chronic cutaneous lupus),包括典型盤狀皮疹(侷限型 [頸部以上]、泛發型 [頸部以上及以下])、肥厚型(疣狀)狼瘡、狼瘡脂膜炎(深部 profundus)、黏膜狼瘡、腫脹性紅斑性狼瘡、凍瘡狼瘡、盤狀狼瘡/扁平苔癬重疊。
- 口腔潰瘍 (Oral ulcers):腭部、頰部、舌部或鼻部潰瘍(在無其他已知原因,如原發性血管炎、感染或糖尿病之情況下)。
- 非瘢痕性禿髮 (Nonscarring alopecia)(瀰漫性稀疏或頭髮脆弱並有可見的斷髮)(在無其他原因,如圓禿、藥物、缺鐵與雄性禿之情況下)。
- 滑膜炎 (Synovitis),侵犯兩個或以上關節,特徵為腫脹或積液,或兩個或以上關節壓痛並有 30 分鐘或以上的晨間僵硬。
- 漿膜炎 (Serositis):典型胸膜炎超過 1 天或胸膜積液或胸膜摩擦音;典型心包性疼痛(仰臥時疼痛,坐起前傾時改善)超過 1 天或心包積液或心包摩擦音或心電圖 (EKG) 顯示心包炎(在無其他原因,如感染、尿毒症與 Dressler 心包炎之情況下)。
- 腎臟 (Renal):尿蛋白/肌酐(或 24 小時尿蛋白)相當於 500 mg 蛋白/24 小時,或紅血球圓柱。
- 神經 (Neurologic):癲癇發作;精神病;多發性單神經炎 (mononeuritis multiplex)(在無其他已知原因,如原發性血管炎之情況下);脊髓炎 (myelitis);周邊或顱神經病變(在無其他已知原因,如原發性血管炎、感染或糖尿病之情況下);急性意識混亂狀態 (acute confusional state)(在無其他原因,包括毒性代謝、尿毒症、藥物之情況下)。
- 溶血性貧血 (Hemolytic anemia)。
- 白血球低下(< 4000/mm³ 至少一次)(在無其他已知原因,如 Felty 症候群、藥物與門脈高壓之情況下),或淋巴球低下(< 1000/mm³ 至少一次)(在無其他已知原因,如 corticosteroid、藥物與感染之情況下)。
- 血小板低下(< 100 000/mm³ 至少一次)(在無其他已知原因,如藥物、門脈高壓與 TTP 之情況下)。
免疫學標準 (Immunological criteria):
- ANA 高於實驗室參考範圍。
- Anti-dsDNA 高於實驗室參考範圍,但 ELISA 例外:為實驗室參考範圍的兩倍以上。
- Anti-Sm。
- 抗磷脂抗體 (Antiphospholipid antibody):下列任一項——狼瘡抗凝血劑 (lupus anticoagulant);梅毒血清反應 (RPR) 偽陽性;中或高效價抗心磷脂抗體 (anticardiolipin)(IgA、IgG 或 IgM);anti-beta 2 glycoprotein I(IgA、IgG 或 IgM)。
- 低補體 (Low complement):低 C3、低 C4、低 CH50。
- 直接 Coombs 試驗 (Direct Coombs test)(在無溶血性貧血之情況下)。
*標準為累積性,無需同時存在。經許可重製自 Petri M., Orbai A.M., Alarcon G.S., et al. Derivation and validation of systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 64, 2012; 2677–2686。
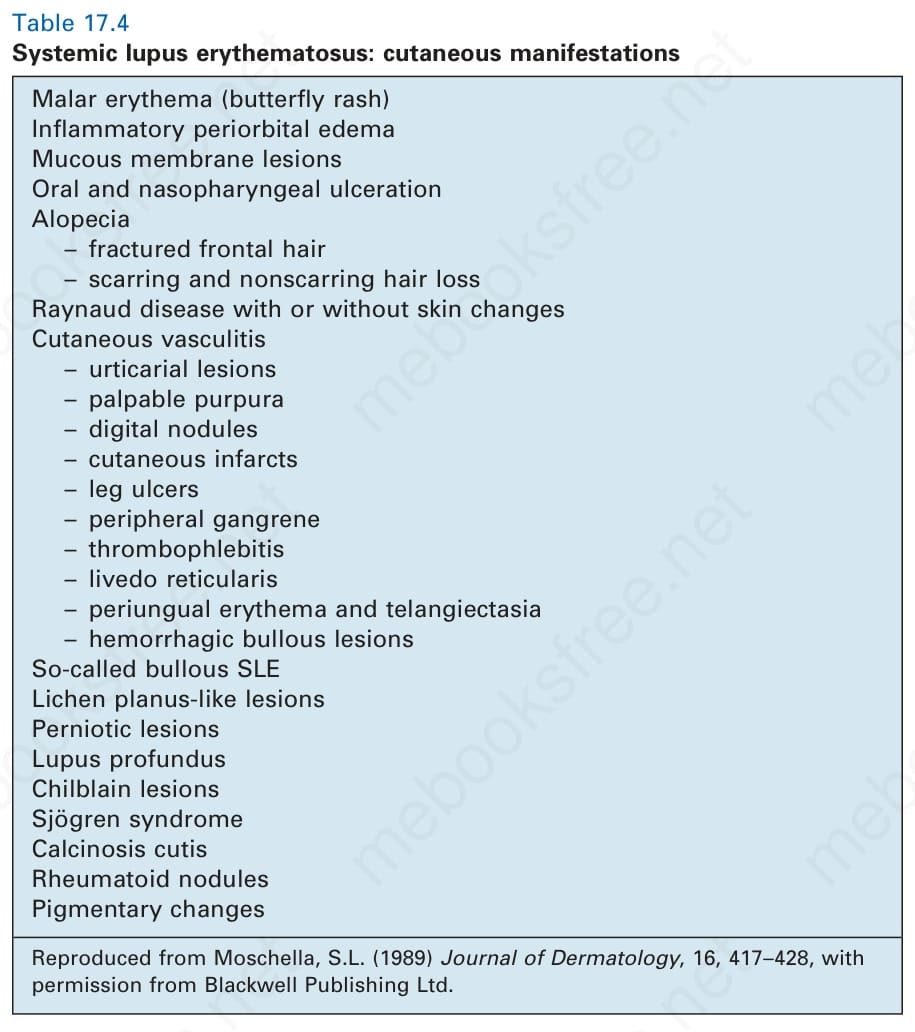
表 17-4:全身性紅斑性狼瘡——皮膚表現 (cutaneous manifestations)。
Table 17.4 Systemic lupus erythematosus: cutaneous manifestations
SLE 的皮膚表現包括:顴部紅斑(蝴蝶斑 butterfly rash);發炎性眶周水腫;黏膜病灶;口腔與鼻咽潰瘍;禿髮(額部頭髮斷裂、瘢痕性與非瘢痕性掉髮);Raynaud 病(伴或不伴皮膚變化);皮膚血管炎(蕁麻疹性病灶、可觸性紫斑、指部結節、皮膚梗塞、腿部潰瘍、周邊壞疽、血栓性靜脈炎、網狀青斑、甲周紅斑與毛細血管擴張、出血性大疱病灶);所謂的大疱型 SLE;扁平苔癬樣病灶;凍瘡樣病灶;深部狼瘡 (lupus profundus);凍瘡病灶;Sjögren 症候群;皮膚鈣質沉著 (calcinosis cutis);類風濕結節;色素變化。
經許可重製自 Moschella, S.L. (1989) Journal of Dermatology, 16, 417–428,Blackwell Publishing Ltd。
中樞神經系統侵犯是罹病與死亡的重要原因,可能影響多達 40% 的病人。腦炎、腦膜炎、血管炎與凝血缺陷,可引起範圍廣泛的臨床表現,包括抽搐、偏癱、舞蹈症 (chorea) 與精神病。抽搐與昏迷是嚴重侵犯的指標,並預示嚴重的結局。周邊神經炎影響多達 12% 的病人;眼部病灶,包括結膜炎、眼底出血與棉絮狀滲出物 (cotton wool exudates),發生於 25%。
腎臟表現發展於約 45% 的病人,進行性腎臟侵犯是罹病與死亡的重要原因(腎病症候群與狼瘡腎絲球腎炎 lupus glomerulonephritis)。活動性腎臟疾病的證據包括尿中每高倍視野超過 5 個紅血球的存在。亦可偵測到大於 1 g/24 hours 的蛋白尿、卵圓形脂肪小體 (oval fat bodies),以及顆粒狀、透明與紅血球圓柱。
泛發性淋巴結病變存在於約 50% 的病人,肝腫大於 20%,脾腫大於 10%。
腸胃道表現不常見;最重要的是食道侵犯,導致蠕動喪失與擴張,令人聯想到硬皮病所見的情形。
實驗室檢查常顯示貧血、白血球低下、淋巴球低下、血小板低下與 ESR 上升。對 reagin 與梅毒螺旋體檢驗呈偽陽性反應常見,10% 至 20% 的病人 Coombs 試驗與類風濕因子陽性,10% 至 50% 有循環抗凝血劑。
雖然紅斑性狼瘡 (LE) 細胞製備曾經是 SLE 的基本篩檢試驗,但現已被抗核因子檢測所取代。此項與其他自體抗體將在致病機轉部分進一步討論。活動性疾病的病人血清補體濃度常偏低(CH50 與 C3);C3 濃度的估計在追蹤疾病活動度上特別有價值。
SLE 的特徵為復發,緩解期長短不一,有時可長達十年或以上。然而,仍有顯著的死亡率。死亡原因包括腎炎、感染與中樞神經系統侵犯。
SLE 與遺傳性補體缺乏之間的關聯已被描述,涉及典型路徑的早期成分,包括 C1r、C1s、C1q、C4 與 C2。C2 缺乏以自體隱性方式遺傳;多達 60% 的同型合子會發展出紅斑性狼瘡,特徵為紅斑性、丘疹鱗屑性、SCLE 樣的光敏性皮膚病、低腎臟侵犯發生率與關節痛。額外特徵可能包括蕁麻疹性血管炎、顴部紅斑與甲褶異常。有時可見盤狀病灶,最近曾記載一位 C2 與 C4 缺乏且合併 LE 脂膜炎的病人。然而,病人通常(但非絕對)抗核因子陰性,且在未受侵犯的皮膚上顯示陰性的狼瘡帶試驗 (lupus band test)。與 HLA-DR2 有關聯,超過 60% 的病人擁有 anti-Ro 抗體。
許多藥物被認定與 SLE 的誘發有關,包括:isoniazid、hydralazine、procainamide、rifampicin、quinidine、penicillamine、terbinafine、carbamazepine、phenytoin、sertraline、valpromide、amiodarone、atenolol、sulfonamides、methimazole、COL-3、hydrochlorothiazide、minocycline、spironolactone、captopril、methyldopa、gold salts、penicillin、streptomycin、phenylbutazone、reserpine、griseofulvin、clonidine、口服避孕藥、captopril、interleukin (IL)-2、hydroxyurea、clobazam、clozapine、ciprofloxacin、cefuroxime、nafcillin、celiprolol、hydralazine、para-amino salicylic acid、yohimbine、infliximab、adalimumab、pegylated interferon alpha-2B、conjugated estrogens、抗腫瘤壞死因子α (anti-TNF-α),以及 etanercept。
短暫性的 SLE 皮膚與血清學表現曾記載於兩名感染 trichophyton mentagrophytes 的兒童。此病也曾與 B 型肝炎疫苗接種以及殺蟲劑暴露有所連結。
Sjögren 症候群共存於約 10% 至 20% 的 SLE 病人。SLE 也曾被描述與下列情況相關:硬皮病、硬斑病、類風濕性關節炎、嗜伊紅性筋膜炎 (eosinophilic fasciitis)、皮肌炎、硬化性苔癬 (lichen sclerosus)、天疱瘡(包括尋常性天疱瘡、落葉狀天疱瘡與副腫瘤性天疱瘡)、Waldenström 高γ球蛋白血症、疱疹樣皮膚炎、潰瘍性結腸炎、圓禿、自體免疫性甲狀腺炎、重症肌無力、黑色棘皮症 (acanthosis nigricans)、Sweet 症候群、紫質症、痛風、類肉瘤病 (sarcoidosis)、乾癬、扁平苔癬,以及皮膚 T 細胞淋巴瘤。除了那些具有自體免疫基礎的疾病外,這些關聯中有許多很可能是偶然的關聯。
新生兒紅斑性狼瘡 (Neonatal lupus erythematosus)
新生兒紅斑性狼瘡 (neonatal lupus erythematosus) 非常罕見,發生率約為每 20 000 名活產嬰兒中 1 例,最常見於女嬰。它與母體 anti-Ro (SS-A) 抗體(95%)與/或 anti-La (SS-B) 抗體相關。也可能存在 anti-U1-ribonucleoprotein (RNP) 抗體。抗心磷脂抗體曾在單一案例中被描述。anti-Ro 與 anti-La 抗體經胎盤轉移,造成一種 SCLE 樣皮疹,由紅斑性環狀鱗屑斑塊組成,特別侵犯眶周區域(「貓頭鷹眼」‘owl-eye’ 或「眼罩」‘eye-mask’)與頭皮,較少侵犯軀幹與四肢 (Figs 17.36 and 17.37)。皮膚變化在出生時即可能存在。儘管如此,它們典型上在嬰兒 4 至 6 週齡時發展,通常為自限性,多數案例在 15 至 17 週時消退而無任何後遺症。這些皮膚病灶常在暴露於紫外線後出現。新生兒紅斑性狼瘡病人的光敏性似乎呈現種族差異,在白種人與美國人中較日本嬰兒更頻繁。結痂病灶與先天性毛細血管擴張性大理石樣皮膚 (cutis marmorata telangiectasia congenita) 是某些案例的額外特徵。例外情況下,也可能發生盤狀病灶、脂膜炎(深部狼瘡 lupus profundus)、多發性硬斑病、糜爛、靶狀病灶與禿髮。萎縮與瘢痕非常罕見,但約 25% 的病人有殘餘的色素減少與毛細血管擴張。後續妊娠所生的嬰兒可能呈現此病的表現,新生兒狼瘡相關心臟疾病的整體復發率為 17%。
新生兒紅斑性狼瘡常伴隨心臟異常。完全性心臟傳導阻斷 (complete heart block) 是最嚴重的併發症,可在子宮內或出生後發展。曾在單一新生兒紅斑性狼瘡相關擴張型心肌病變的病人偵測到對 annexin A6 的循環自體抗體。與心臟侵犯相關的死亡率約為 19%。血液學併發症估計發展於 10% 至 35% 的新生兒狼瘡病人;最常見的異常為輕度貧血與輕度血小板低下。曾在單一病人報告多發性皮膚黏膜與內臟血管瘤。無皮膚或心臟侵犯證據的嚴重血液學與肝臟侵犯曾例外性報告。新生兒狼瘡的肝臟侵犯罕見,通常表現為膽汁鬱積性肝炎。其他罕見關聯包括伴有直腸出血的乙狀結腸毛細血管擴張、疼痛性足底萎縮、中樞神經系統血管炎、中風、腦室內出血,以及廣泛性主動脈瘤。
嬰兒體內的母體自體抗體濃度在生命最初數週迅速下降,一般在 1 歲時變得無法偵測。
此病也發生於同卵與異卵雙胞胎。曾描述單一案例與 Turner 症候群相關。存活至嬰兒期的新生兒預後相當良好,但因心臟疾病,整體死亡率約為 10%。受影響嬰兒的母親初期常無症狀,但她們可能患有全身性狼瘡、Sjögren 症候群、類風濕性關節炎、白血球碎裂性血管炎或重疊症候群。
許多初期無症狀的母親,常隨後發展出結締組織疾病的證據,特別是全身性狼瘡(約 20% 的案例)與 Sjögren 症候群。
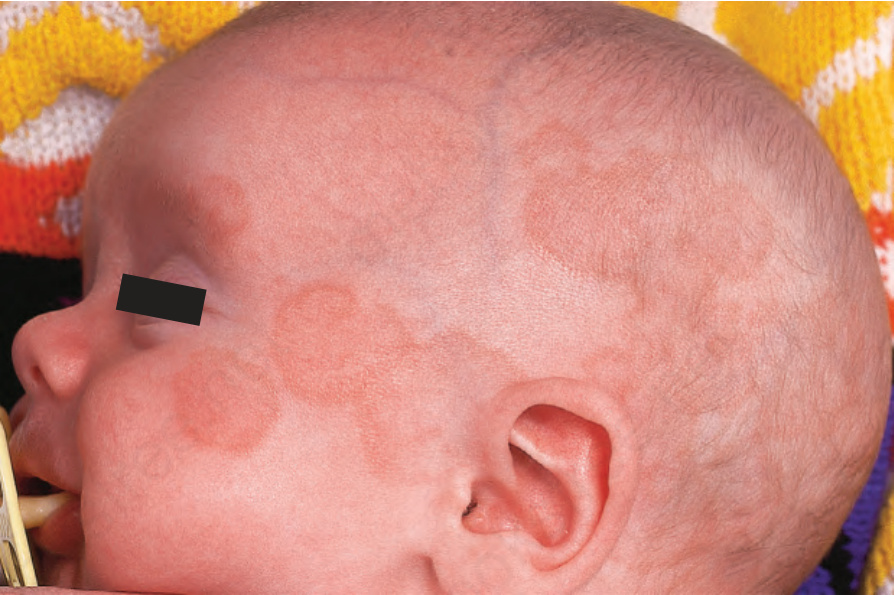
圖 17-36:新生兒紅斑性狼瘡——注意臉頰、前額與頭皮上紅斑性、略有鱗屑的斑塊。
Fig. 17.36 Neonatal lupus erythematosus: note the presence of erythematous, slightly scaly plaques on the cheeks, forehead, and scalp. By courtesy of the Institute of Dermatology, London, UK.
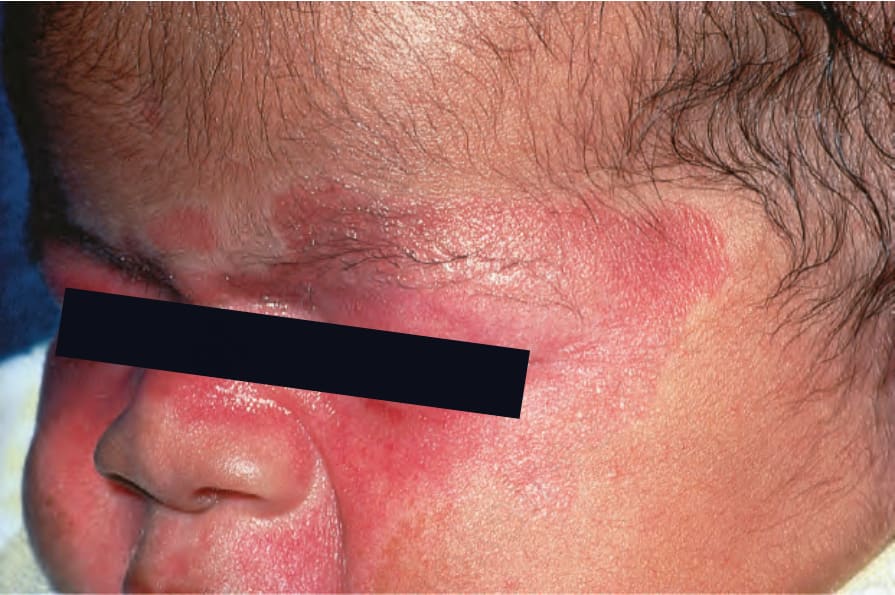
圖 17-37:新生兒紅斑性狼瘡——此嬰兒臉頰、鼻部與眼周有強烈的紅斑。
Fig. 17.37 Neonatal lupus erythematosus: in this child, there is intense erythema affecting the cheeks and nose and around the eyes.
致病機轉與組織學特徵 (Pathogenesis and Histologic Features)
儘管投入龐大的研究努力,確切的病因與致病機轉仍屬未知,但此病確實是多因子的。紅斑性狼瘡的特徵為 B 細胞過度活躍,並伴隨抑制性 T 細胞功能缺陷。病人發展出範圍廣泛的自體抗體,其中許多形成免疫複合體並造成全身性表現,包括血管炎與腎絲球腎炎。除了免疫學因子外,家族、遺傳與荷爾蒙影響也扮演角色。一般相信在紅斑性狼瘡中存在遺傳易感性;性別相關與環境因子是促進疾病發展所必需。
自體抗體在紅斑性狼瘡的診斷中很重要,並在其致病機轉中具有顯著作用,可透過直接細胞毒性效應(例如抗淋巴球抗體所誘發的淋巴球低下)或免疫複合體沉積而起作用。直到 1960 年代,病人血液中 LE 細胞的存在被視為紅斑性狼瘡的病理特徵 (pathognomonic)。然而,這一觀念因以直接免疫螢光技術發現抗核因子(抗核抗體)而改變。抗核抗體存在於 90% 至 95% 的 SLE 病人、30% 的侷限型 DLE 病人,以及 50% 的泛發型 DLE 病人血清中。亦應注意,10% 的正常人口血清中有抗核抗體,惟濃度偏低。抗核抗體至少有四種亞型:
- 同質型或瀰漫型 (homogeneous or diffuse pattern) 最常見 (Figs 17.38 and 17.39)。
- 斑點型螢光 (Speckled fluorescence) 代表對鹽水可溶性核蛋白之抗體,存在於紅斑性狼瘡-多形性紅斑症候群病人 (Fig. 17.40)。
- 核仁型 (nucleolar pattern,代表抗核仁 RNA 抗體) 偶爾在紅斑性狼瘡中可見,雖然它在系統性硬化症 (systemic sclerosis) 中更常見。
- 周邊(輪廓)染色型 (peripheral [outline] staining pattern) 反映高效價 anti-DNA 抗體,是活動性全身性疾病的標記。
紅斑性狼瘡病人發展出對多種核抗原與細胞質抗原的抗體;這些自體抗體,無論單獨或合併出現,皆有不同的關聯性,可允許(至少在某種程度上)預測個別病人的疾病病程 (Table 17.5)。對 native(雙股)DNA (nDNA) 之抗體是特發性(典型)SLE 的病理特徵 (Fig. 17.41)。它們在藥物誘發型變異型中罕見,僅偶爾出現於 DLE 病人。anti-nDNA 抗體與低補體血症 (hypocomplementemia) 並存時,是活動性疾病的指標,並常伴隨嚴重的腎臟侵犯。
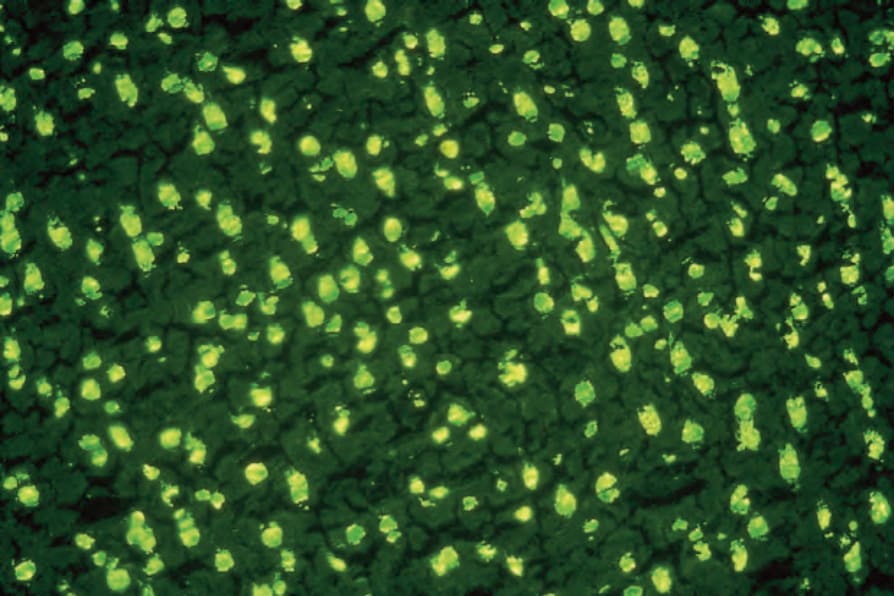
圖 17-38:全身性紅斑性狼瘡——大鼠肝臟中的同質型抗核抗體。
Fig. 17.38 Systemic lupus erythematosus: homogeneous antinuclear antibody in rat liver. By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
圖 17-39:全身性紅斑性狼瘡——高倍視野。
Fig. 17.39 Systemic lupus erythematosus: high-power view. By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
圖 17-40:全身性紅斑性狼瘡——斑點型抗核抗體(HEP II)。
Fig. 17.40 Systemic lupus erythematosus: speckled antinuclear antibody – HEP II. By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
圖 17-41:全身性紅斑性狼瘡——anti-dsDNA(Crithidia luciliae)。
Fig. 17.41 Systemic lupus erythematosus: anti-dsDNA (Crithidia luciliae). By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
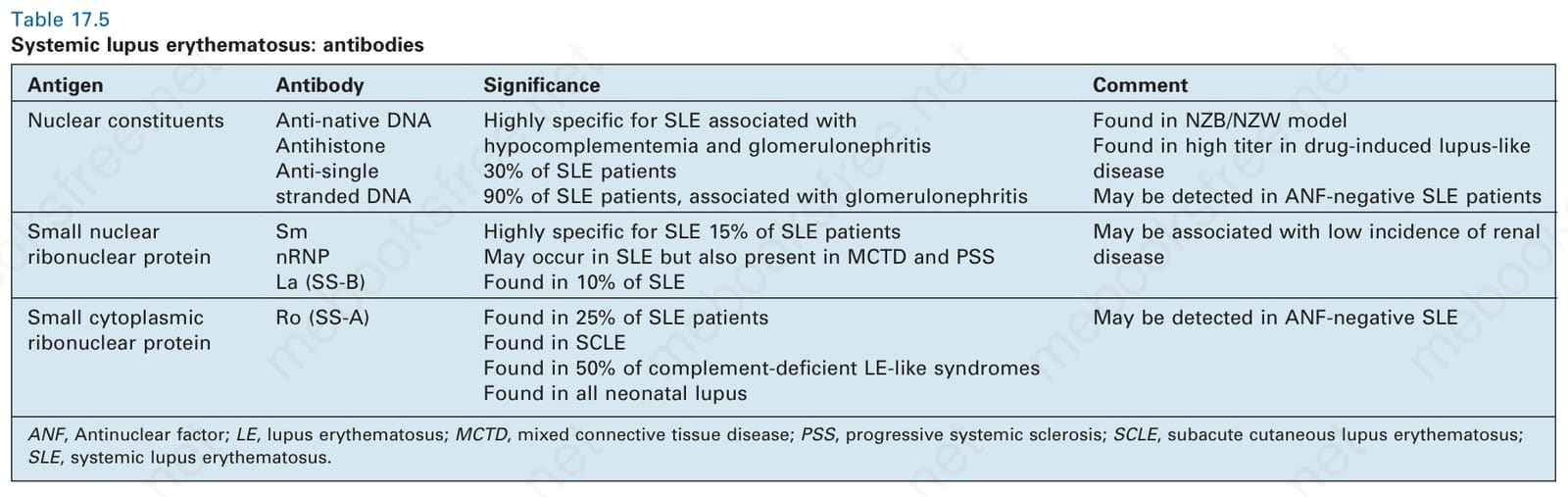
表 17-5:全身性紅斑性狼瘡——抗體 (antibodies)。
Table 17.5 Systemic lupus erythematosus: antibodies
此表整理 SLE 的抗原、抗體與意義/註解。核成分 (Nuclear constituents):Anti-native DNA——對 SLE 高度專一,與低補體血症及腎絲球腎炎相關;Antihistone——見於 30% 的 SLE 病人,在 NZB/NZW 模型中發現,在藥物誘發性狼瘡樣疾病中以高效價出現;Anti-single stranded DNA——見於 90% 的 SLE 病人,與腎絲球腎炎相關,可在 ANF 陰性的 SLE 病人偵測到。小核核糖核蛋白 (Small nuclear ribonuclear protein):Sm——對 SLE 高度專一,見於 15% 的 SLE 病人;nRNP——可發生於 SLE,但也存在於 MCTD 與 PSS;La (SS-B)——見於 10% 的 SLE,可在 ANF 陰性的 SLE 病人偵測到。小細胞質核糖核蛋白 (Small cytoplasmic ribonuclear protein):Ro (SS-A)——見於 25% 的 SLE 病人、見於 SCLE、見於 50% 的補體缺乏性 LE 樣症候群、見於所有新生兒狼瘡,可能與低腎臟疾病發生率相關。
ANF,抗核因子;LE,紅斑性狼瘡;MCTD,混合型結締組織疾病;PSS,進行性系統性硬化症;SCLE,亞急性皮膚紅斑性狼瘡;SLE,全身性紅斑性狼瘡。
抗組織蛋白抗體 (Antihistone antibodies) 可在約 30% 的特發性 SLE 病人中發現,但特別與藥物誘發型變異型相關。後者由多種藥物引起,包括 hydralazine、procainamide hydrochloride、phenytoin 與 isoniazid。多達 20% 服用 procainamide 的病人會發展出症狀,50% 出現抗核抗體。procainamide 誘發的 SLE 樣症候群以白血球專一性抗核抗體之存在為特徵。臨床特徵包括倦怠、伴有胸膜積液的肺炎、關節痛、關節炎與漿膜炎;腎臟、中樞神經系統與皮膚病灶較不常見,且通常輕微。雖然抗核抗體常存在,但多數藥物誘發性狼瘡案例不形成 anti-DNA 抗體。藥物誘發性狼瘡通常(但非絕對)在停藥後緩解。
對 ssDNA 之抗體出現於 90% 的典型 SLE 病人,以及約 20% 的播散型 DLE 病人,也可在其他結締組織疾病中發現。它們在播散型 DLE 中的存在與發展為 SLE 的風險增加相關。抗核抗體陰性的 SLE 病人,其血清中可能有 anti-ssDNA 抗體。對可溶性核抗原 Sm 之抗體對 SLE 高度專一。它們也可能在一群預後良好的病人中發現,這些病人抗核因子陽性、但 anti-nDNA 陰性,並有輕度非進行性腎絲球腎炎、低補體血症、輕度中樞神經系統侵犯與顯著的皮膚皮疹。
對核糖核蛋白 (ribonucleoprotein) 之抗體可在 23% 的 SLE 病人中偵測到,但更常與混合型結締組織疾病 (mixed connective tissue disease, MCTD) 相關:它們也可在進行性系統性硬化症病人中發現。anti-La (SS-B) 抗體可在 10% 的 SLE 病人血清中偵測到。anti-La 抗體常存在於 Sjögren 症候群,幾乎總是伴隨 anti-Ro (SS-A) 抗體。後者特別與 SCLE、補體缺乏性紅斑性狼瘡、循環 IgG 或 IgM 抗凝血劑,以及新生兒紅斑性狼瘡相關。約 15% 至 20% 的 SLE 病人有可偵測到的抗嗜中性球細胞質抗體 (antineutrophil cytoplasmic antibodies, ANCA)。一項涵蓋 645 名巴西兒童 SLE 病人的大型多中心研究顯示,anti-Ro (SS-A) 與/或 anti-La (SS-B) 抗體與輕度皮膚及肌肉骨骼侵犯相關。
循環抗凝血劑(抗磷脂或狼瘡抗凝血劑,antiphospholipid or lupus anticoagulant)與下列情況相關:矛盾性血栓 (paradoxical thrombosis)、自發性流產、早產、子宮內死亡、不穩定型高血壓、皮膚壞死、壞疽、瘀斑、紫斑、腿部潰瘍、白色萎縮、網狀青斑,以及偽陽性梅毒血清學——即狼瘡抗凝血劑症候群 (lupus anticoagulant syndrome) (Table 17.6)。狼瘡抗凝血劑症候群更準確地稱為抗磷脂症候群 (antiphospholipid syndrome),因為並非所有病人都伴有 SLE。它源於循環抗磷脂抗體的存在,這些抗體在體外 (in vitro) 抑制凝血,但更重要的是,與影響動脈與靜脈之血栓現象大幅增加的風險相關 (Figs 17.42 and 17.43)。約 50% 的 SLE 病人呈現抗磷脂抗體,但這些病人中只有一半會發展出疾病表現。皮膚上的表現也可能為丘疹與結節,或類似壞疽性膿皮症 (pyoderma gangrenosum) 的病灶。有趣的是,具有抗磷脂抗體與 SLE 的病人似乎較有發展為皮膚鬆弛症 (anetoderma) 的風險。然而,其發生率並不高。有趣的是,具有臨床顯著抗磷脂抗體的病人,與無顯著抗磷脂抗體的病人相比,被發現有較低的急性皮膚狼瘡盛行率。罕見情況下,病人發展出反應性血管內皮瘤病 (reactive angioendotheliomatosis)。全身性侵犯包括深部靜脈血栓,常併發肺栓塞、動脈阻塞並造成中風、暫時性腦缺血發作、多發梗塞性失智、心肌梗塞與壞疽。
皮膚血栓病灶、分枝狀青斑 (livedo racemosa)、高血壓與腦血管疾病的關聯被稱為 Sneddon 症候群 (Sneddon syndrome)。此症主要表現於年輕女性,在兒童中屬例外。約 41% 的病人有抗磷脂抗體。
抗磷脂抗體如何誘發血栓尚屬未知。抑制 protein C(一種天然的維生素 K 依賴性抗凝血劑)功能的理論,以及對內皮細胞 prostacyclin 的抑制效應,皆尚未被證實。抗磷脂症候群也曾被記載於其他自體免疫疾病,包括類風濕性關節炎、溶血性貧血、血小板低下性紫斑、潰瘍性結腸炎、factor V Leiden 突變與間皮瘤。它也可能併發於數種藥物治療,包括 phenothiazines 與 procainamide,可在病毒性疾病期間發展,包括後天免疫缺乏症候群 (AIDS),並可伴隨潛在的淋巴瘤表現。抗磷脂症候群於他處有進一步討論。
anti-Ro 抗體已被證明在新生兒與亞急性紅斑性狼瘡中結合至表皮,並在新生兒紅斑性狼瘡中結合至傳導系統與心肌。它們也在體外 (in vitro) 結合至角質形成細胞,且當灌注入移植人類皮膚的小鼠時,已被證明會與表皮反應。此一發現——結合隨母體 IgG 從循環中移除而皮膚表現消失的現象——提示在新生兒紅斑性狼瘡中,anti-Ro 抗體具有重大致病意義。Ro 抗原存在於角質形成細胞的核與細胞質中,且已證明 UVB 能夠將這些抗原轉位至培養的角質形成細胞表面。血清中的 anti-Ro 抗體結合至角質形成細胞中的抗原,似乎在誘發抗體依賴性角質形成細胞損傷上很重要。
anti-C1q 抗體存在於約 30% 的 SLE 病人。anti-C1q 抗體、anti-dsDNA 抗體與低補體的組合,已被證明是狼瘡腎炎最可靠的血清學標記。
免疫複合體已被證明在 SLE 之血管炎與腎絲球腎炎的致病機轉中很重要;其存在可藉由在皮膚切片之血管壁中偵測到免疫球蛋白與補體而推斷 (Fig. 17.44)。在狼瘡腎絲球腎炎病人的腎皮質中,已偵測到高濃度的 anti-nDNA、anti-ssDNA 與 anti-Ro (SS-A) 抗體。在皮膚病灶的真皮浸潤中發現大量 T 淋巴球與巨噬細胞,而 B 細胞極少或無,提示細胞媒介免疫 (cell-mediated immunity) 可能在此部位病灶的致病機轉中特別重要。遲發型過敏反應與抗體依賴性細胞毒性兩種機轉皆曾被提出。
SLE 中存在顯著的女性優勢,提示女性性荷爾蒙具有致病意義。有趣的是,在兔的 SLE 實驗模型中,雌性比雄性有嚴重得多的腎絲球腎炎,而此差異可藉由閹割或投予雄性性荷爾蒙而抵消。
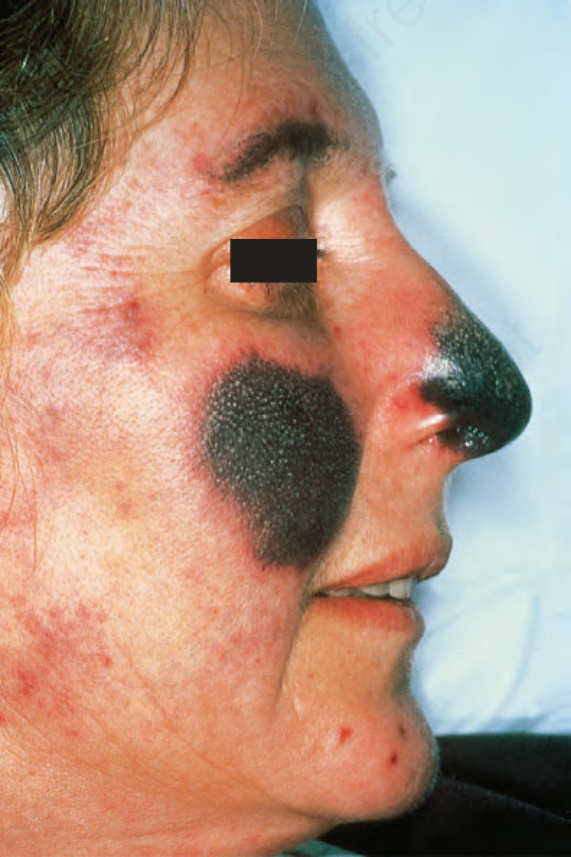
圖 17-42:狼瘡抗凝血劑症候群——廣泛壞疽伴潰瘍,侵犯鼻部與臉頰。
Fig. 17.42 Lupus anticoagulant syndrome: extensive gangrene with ulceration affecting the nose and cheek. By courtesy of C. Stephens, MD, Poole Hospital, Poole, UK.
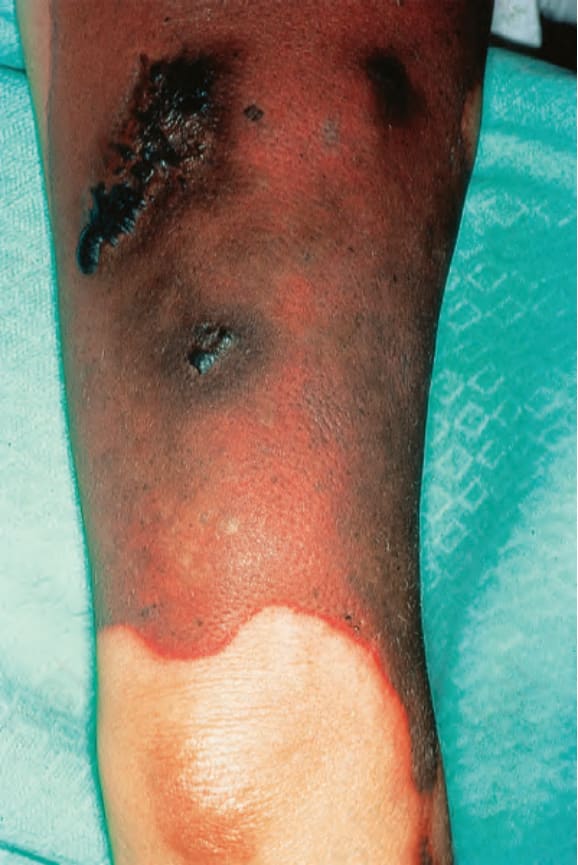
圖 17-43:狼瘡抗凝血劑症候群——腿部也受侵犯。
Fig. 17.43 Lupus anticoagulant syndrome: the leg was also involved. By courtesy of C. Stephens, MD, Poole Hospital, Poole, UK.
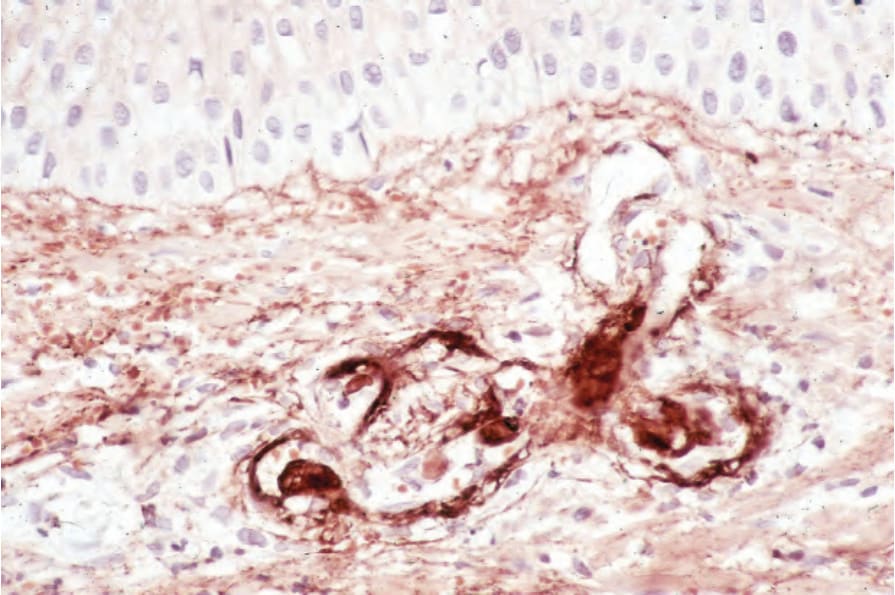
圖 17-44:全身性紅斑性狼瘡——免疫過氧化酶反應 (immunoperoxidase reaction) 顯示血管壁內的 IgG。
Fig. 17.44 Systemic lupus erythematosus: immunoperoxidase reaction demonstrating IgG within a blood vessel wall.
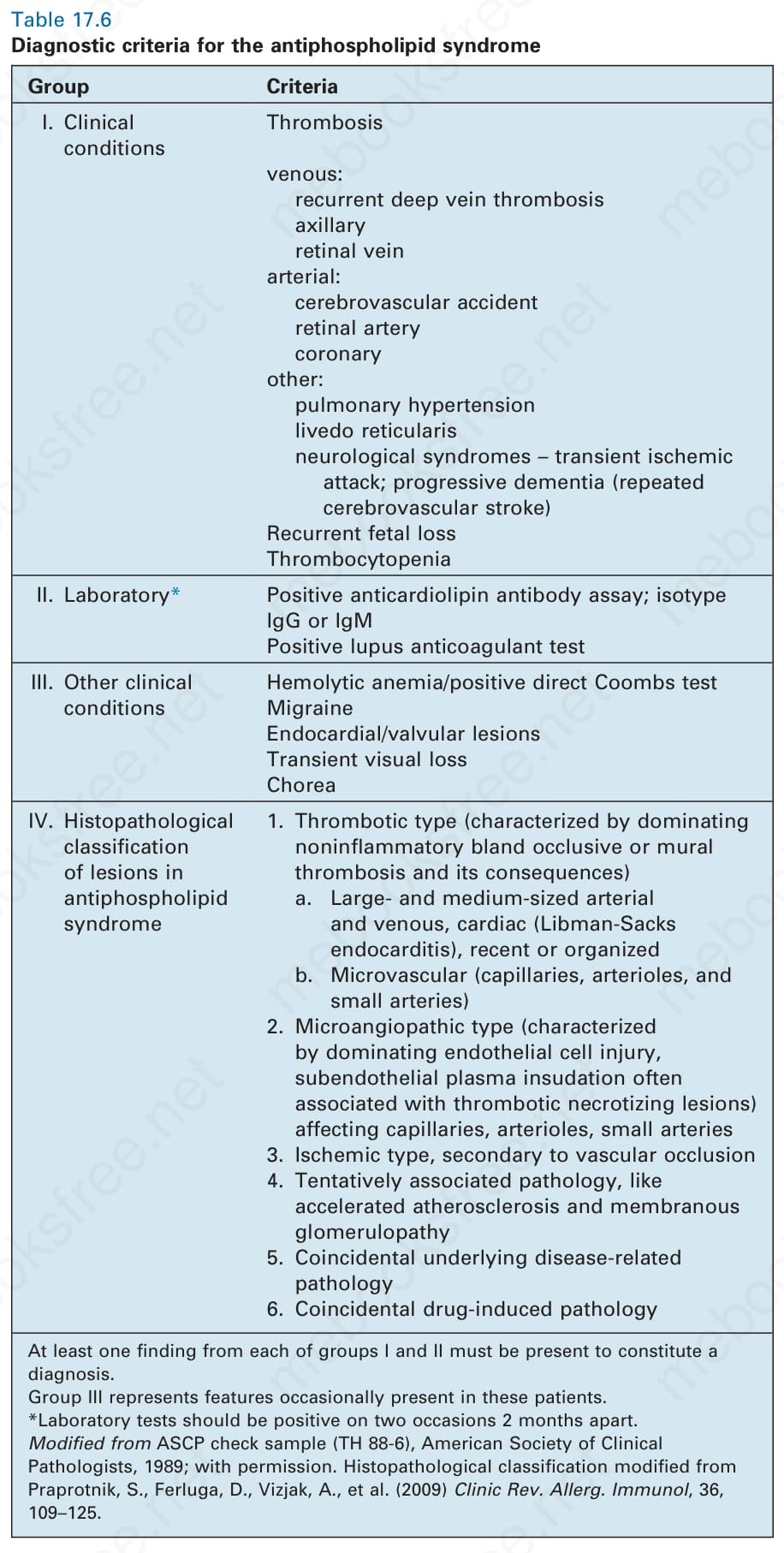
表 17-6:抗磷脂症候群的診斷標準 (diagnostic criteria for the antiphospholipid syndrome)。
Table 17.6 Diagnostic criteria for the antiphospholipid syndrome
抗磷脂症候群的診斷標準分組如下。
第 I 組 臨床 (Clinical):血栓情況——靜脈:反覆深部靜脈血栓、腋靜脈、視網膜靜脈;動脈:腦血管意外、視網膜動脈、冠狀動脈;其他:肺高壓、網狀青斑、神經症候群(暫時性腦缺血發作;進行性失智 [反覆腦血管中風]);反覆胎兒流失;血小板低下。
第 II 組 實驗室 (Laboratory)*:抗心磷脂抗體檢測陽性;同型 IgG 或 IgM;狼瘡抗凝血劑試驗陽性。
第 III 組 其他臨床情況 (Other clinical conditions):溶血性貧血/直接 Coombs 試驗陽性;偏頭痛;心內膜/瓣膜病灶;暫時性視力喪失;舞蹈症。
第 IV 組 抗磷脂症候群病灶之組織病理學分類 (Histopathological classification of lesions):
- 血栓型 (Thrombotic type,以主導性的非發炎性、平淡的阻塞性或壁性血栓及其後果為特徵):a. 大型與中型動脈與靜脈、心臟(Libman-Sacks 心內膜炎)、近期或機化的;b. 微血管(毛細血管、小動脈與小動脈分支)。
- 微血管病型 (Microangiopathic type,以主導性的內皮細胞損傷、內皮下血漿滲入並常伴血栓性壞死病灶為特徵),侵犯毛細血管、小動脈、小動脈分支。
- 缺血型 (Ischemic type),繼發於血管阻塞。
- 暫時性相關病理,如加速性動脈粥樣硬化與膜性腎絲球病變。
- 巧合的潛在疾病相關病理。
- 巧合的藥物誘發病理。
第 I 組與第 II 組各須至少有一項發現,方能構成診斷。第 III 組代表這些病人偶爾出現的特徵。*實驗室檢驗應於相隔 2 個月的兩次場合呈陽性。修改自 ASCP check sample (TH 88-6), American Society of Clinical Pathologists, 1989。組織病理學分類修改自 Praprotnik, S., Ferluga, D., Vizjak, A., et al. (2009) Clinic Rev. Allerg. Immunol, 36, 109–125。
無疑地,SLE 的發展存在遺傳易感性。有許多家族性發生的例子,且免疫球蛋白異常常被記載於無症狀的親屬中;事實上,單一家族中曾記錄多達七名成員受影響。紅斑性狼瘡也曾被報告於同卵雙胞胎。HLA 分型顯示 SLE 中 HLA-A1、HLA-B8、HLA-B15、HLA-DR2 與 HLA-DR3 的發生率增加。有人提出此病的表現可能以自體顯性性狀遺傳。HLA-DR4 與 hydralazine 誘發的紅斑性狼瘡相關。HLA-DRw6 與 HLA-B8 的頻率在 DLE 中增加。最近已證明,具有多形性日光疹 (polymorphic light eruption) 與 HLA-DRB1*0301 的病人,發展為亞急性或盤狀紅斑性狼瘡的風險較高。多形性日光疹在紅斑性狼瘡病人親屬中的家族聚集現象,提示這些疾病可能共享相似的致病機轉。
SCLE 已被證明與 TNF-308A 等位基因相關。SCLE 與 TNF-alpha (TNF-α) 基因啟動子的單核苷酸多態性,以及編碼 C1q 之 C1qA 鏈的基因之間的關聯已被證明。TNF-α 的轉錄在亞急性狼瘡中似乎受光調節。由角質形成細胞所產生的 TNF-α,已被定位於 SCLE 的頑固病灶內,提示其在 SCLE 致病機轉中的潛在角色。CD4+ T 淋巴球中 DNA 甲基化的異常調節,最近被認為與 SCLE 的致病機轉有關。紅斑性狼瘡的觸發機制包括 UV 光(自然發生與人工皆有)、藥物,以及可能的病毒。日光常使皮膚表現惡化,並可能加重全身性疾病。已在 SLE 病人中鑑定出對 UV DNA 之抗體。這些抗體在光敏性誘發病灶的致病機轉中可能很重要。病灶在春夏季典型上較嚴重,且皮膚病灶可由 UV 照射人工誘發。細胞凋亡 (apoptosis) 的失調已被提出為紅斑性狼瘡致病機轉中的一個重要機制。表皮基底細胞中 bcl-2 表現的減少與 FAS 抗原的過度表現相關,而這似乎與表皮中細胞凋亡的程度相關。
儘管早期對紅斑性狼瘡的病毒病因充滿熱情,廣泛的研究並未提供令人信服的證據。多年來,內皮細胞細胞質內的副黏液病毒樣 (paramyxovirus-like) 包涵體曾被認為代表病毒病因的證據。現在已知它們代表一種非特異性的膜性副產物(小管網狀體 tubuloreticular body),由胞器退化所生成。曾有人提出與小病毒(紅病毒 erythrovirus)B19 的關聯。曾描述兩種病毒誘發的實驗動物模型——阿留申貂病 (Aleutian mink disease) 與紐西蘭黑白雜交小鼠病 (New Zealand black–white hybrid mouse disease)。近期的研究顯示,後者與一種原發性幹細胞缺陷相關。因此骨髓移植可將疾病轉移至先前接受照射的正常受贈者,並誘發耐受性缺陷。骨髓培養產生具有增強抗體合成能力的 B 細胞。鼠類狼瘡與其人類對應物呈現顯著的相似性。有趣的是,native(雙股)DNA 抗體在吸菸的 SLE 病人中比不吸菸的 SLE 病人更常被偵測到。事實上,一項涵蓋 405 名紅斑性狼瘡病人的大型研究確認,吸菸、盤狀狼瘡與腫脹性狼瘡之間有高度關聯。
免疫組化與輔助檢查 (Immunofluorescence & Ancillary Studies)
狼瘡帶試驗 (Lupus band test)
狼瘡帶試驗在紅斑性狼瘡的診斷中特別重要。它在較小程度上對評估預後也有一些價值。藉由免疫螢光或免疫過氧化酶技術,在真皮-表皮交界 (dermal–epidermal junction) 處尋找免疫球蛋白與補體的存在 (Fig. 17.45)。最常被鑑定到的是 IgM,雖然 IgG、IgA 與 C3 也常存在。IgG 沉積似乎是對紅斑性狼瘡最專一的。其他可被鑑定的因子包括 properdin、C1q 與 C4。膜攻擊複合體 (membrane attack complex, MAC) 的沉積也已被證明是皮膚紅斑性狼瘡相對敏感且專一的標記。雖然沉積物通常為同質性,但也認可顆粒狀與絲狀(網狀)型態。同質性帶是慢性病灶的典型表現,顆粒狀帶見於未受侵犯的皮膚,絲狀沉積通常是早期病灶的特徵。在超微結構上,免疫反應物存在於基底板下 (sub-basal lamina) 結締組織中,並與重複的緻密板 (reduplicated lamina densa) 密切相關 (Fig. 17.46)。免疫球蛋白沉積不一定與皮膚病灶的臨床或組織學存在相關,因此不太可能具有致病意義。
狼瘡帶試驗在約 50% 至 94% 的 SLE 病人、60% 至 80% 的盤狀變異型病人、60% 的 SCLE 病人,以及 50% 的新生兒紅斑性狼瘡嬰兒之受侵犯皮膚中呈陽性。受侵犯的黏膜也顯示免疫反應物沉積,這在組織學上無法與扁平苔癬區分的病人中可能特別有價值。它在多達 67% 的全身性疾病病人之未受侵犯皮膚中也可能呈陽性。未受侵犯皮膚陽性試驗的盛行率取決於部位,以肩部皮膚的發生率最高(70%)。日曬皮膚,如前臂背側面,比非日曬皮膚(50–60%)更常呈陽性(60–70%)。然而,20% 的健康年輕成人之日曬正常皮膚標本顯示陽性狼瘡帶試驗,這一觀察指出非日曬皮膚才是首選的基質。
陽性也取決於疾病的持續時間(病灶少於 3 個月者常為陰性)與先前類固醇治療的影響。較少見的情況下,在 DLE 病人的未受侵犯皮膚也可見陽性免疫螢光。然而,結果必須在臨床資訊的脈絡中加以解讀。陽性 IgM 狼瘡帶試驗可見於不相關的情況,如日光角化症 (solar keratosis)、多形性日光疹、酒糟、淋巴球浸潤 (lymphocytic infiltrate) 與皮肌炎,以及作為 UV 照射的結果。後述這些情況通常伴隨 C3 沉積或單一免疫球蛋白類別,特別是 IgM,相對於紅斑性狼瘡中所見的多種免疫球蛋白亞類。在非狼瘡情況中,狼瘡帶通常較淡且呈斑塊狀。由於有如此顯著的偽陽性率,須強調狼瘡帶試驗的結果必須相當謹慎地解讀,並始終置於臨床資訊與組織學特徵的脈絡中。
具有 SCLE 的病人曾被描述有一種特徵性的微粒塵狀免疫球蛋白沉積(主要為 IgG),影響表皮的基底細胞。有時基底上表皮、附屬器上皮與真皮細胞浸潤顯示類似的螢光。此沉積型態與 Ro 抗體的存在相關。它偶爾也見於 SLE。在某些案例中,可見角質形成細胞 IgG 的斑點型核染色於結締組織疾病中。在 SLE 的情境下,此發現似乎與較低的腎臟疾病發生率相關。
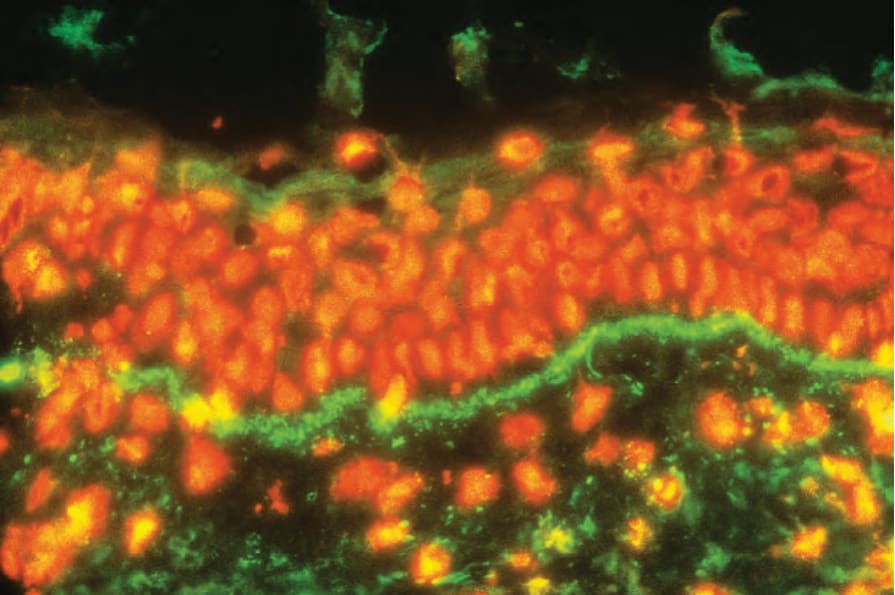
圖 17-45:全身性紅斑性狼瘡——陽性帶試驗 (band test)(IgG)。
Fig. 17.45 Systemic lupus erythematosus: positive band test (IgG). By courtesy of the Department of Immunofluorescence, Institute of Dermatology, London, UK.
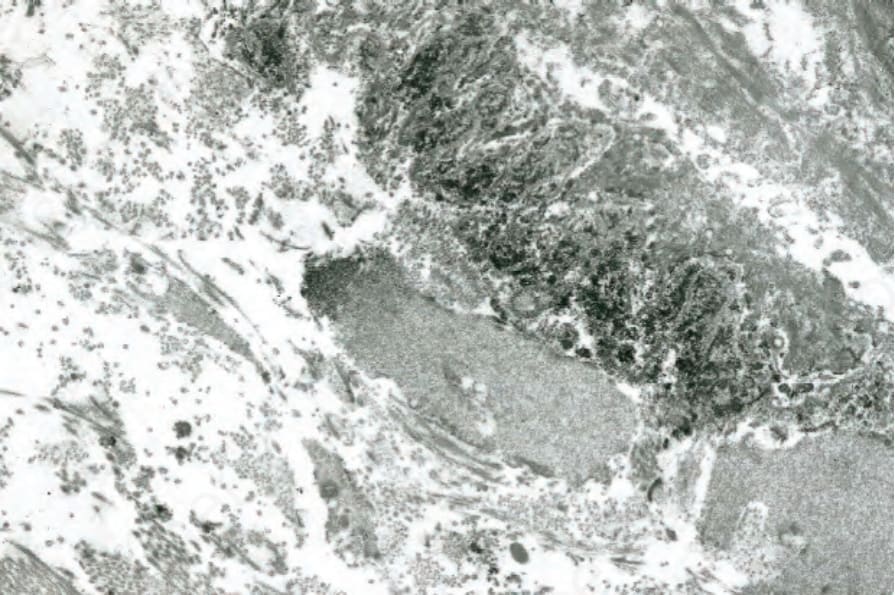
圖 17-46:狼瘡帶試驗——免疫電子顯微鏡(冷凍切片)顯示緻密板 (lamina densa) 下方的 IgM 沉積。
Fig. 17.46 Lupus band test: immunoelectron microscopy (frozen section) showing IgM deposition below the lamina densa.
組織病理特徵 (Histopathology)
各種紅斑性狼瘡亞型的組織病理特徵(深部紅斑性狼瘡與 SLE 的大疱性皮膚病除外)顯示相當大的重疊,以顯微技術區分它們常很困難。
盤狀紅斑性狼瘡 (Discoid lupus erythematosus)
DLE 顯示多變的特徵,取決於疾病的階段。活動性病灶以過度角化與毛囊擴張並伴角質栓塞為特徵;偶爾出現局部角化不全 (parakeratosis) (Figs 17.47–17.49)。表皮通常萎縮且相當扁平,雖然有時有棘層肥厚 (acanthosis) (Fig. 17.50)。在真皮-表皮交界處所見最重要的特徵為:
- 表皮基底層的液化變性 (liquefactive degeneration) (Fig. 17.51),
- 基底膜增厚 (basement membrane thickening) (Fig. 17.52),可藉由 periodic acid-Schiff (PAS) 反應而更加突顯。
這兩個特徵還可能影響毛囊上皮,有時也可見真皮血管的基底膜增厚 (Fig. 17.53)。表皮基底層的液化變性常伴隨色素失禁 (pigmentary incontinence) (Fig. 17.54)。在某些情況下,表皮變化伴隨類膠樣體 (cytoid body) 形成,但這通常不像扁平苔癬中那麼明顯 (Fig. 17.55)。偶爾可見類澱粉 (amyloid) 形成。
真皮乳頭呈水腫性,且常存在毛細血管擴張性血管 (Fig. 17.56)。有時可見紅血球的局部外滲 (Fig. 17.57)。DLE 的特徵是血管周圍與附屬器周圍的慢性發炎細胞浸潤,由淋巴球與數量不等的組織細胞 (histiocytes) 組成 (Figs 17.58 and 17.59)。最常見的細胞是 T 淋巴球,包括輔助與抑制細胞。嗜中性球多形核細胞 (neutrophil polymorphs) 與漿細胞通常不明顯。然而,在一位伴有 X 染色體連鎖慢性肉芽腫病之狼瘡樣皮膚病的病人中,曾描述其狼瘡樣病灶有嗜中性球與白血球碎裂 (leukocytoclasia)。CD4+ 細胞在 DLE 皮膚病灶中占多數。這些 T 淋巴球多數為 Ia+,指示活化狀態。偶有 CD4+ 細胞存在於表皮中,與基底角質形成細胞損傷灶密切相關。表皮 Langerhans 細胞數量通常減少。B 淋巴球一般僅以少量存在。浸潤在真皮乳頭典型上稀疏,但局部緻密的聚集物特徵性地見於網狀真皮 (reticular dermis),有時可延伸至皮下脂肪。
真皮中糖胺聚醣 (glycosaminoglycans,酸性黏多醣 acid mucopolysaccharides) 的增加是急性病灶的常見特徵 (Fig. 17.60);在腫脹性狼瘡中,此現象非常顯著 (Figs 17.61 and 17.62)。然而,最不常見的是,盤狀狼瘡中真皮黏液沉積可能廣泛,臨床上表現為丘疹結節性黏液病 (papulonodular mucinosis)。非常罕見地,曾記載真皮鈣化 (Fig. 17.63)。例外情況下,可見真皮膠原蛋白的「類纖維蛋白」(‘fibrinoid’) 變化 (Fig. 17.64)。
漿細胞樣樹突細胞 (plasmacytoid dendritic cells) 占真皮發炎細胞浸潤超過 10%、其排列成簇,或其存在於真皮-表皮交界處,已被證明在盤狀與疣狀(肥厚型)紅斑性狼瘡中具有顯著的診斷價值。
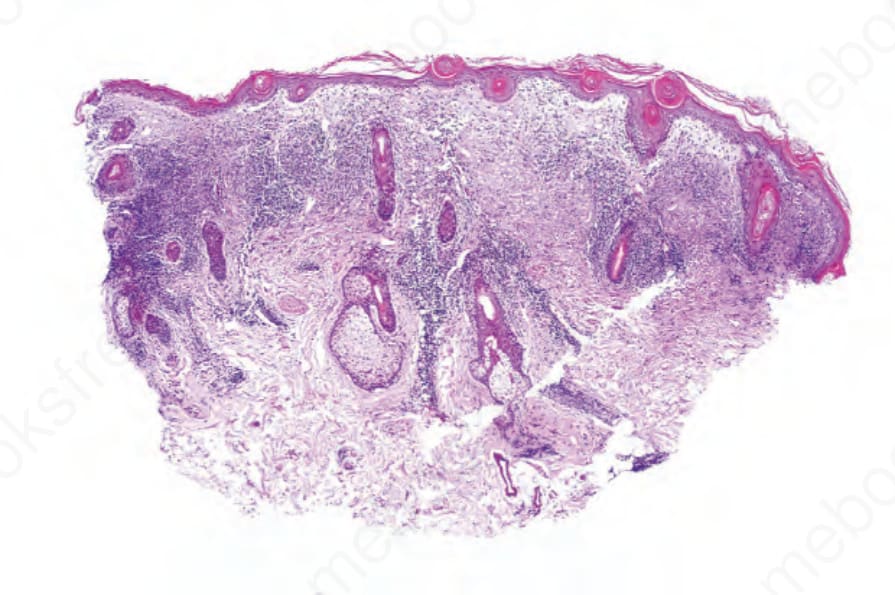
圖 17-47:盤狀紅斑性狼瘡——此掃描視野顯示過度角化、毛囊栓塞、萎縮的表皮,以及血管周圍與附屬器周圍的慢性發炎細胞浸潤。
Fig. 17.47 Discoid lupus erythematosus: this scanning view shows hyperkeratosis, follicular plugging, an atrophic epidermis, and a perivascular and periadnexal chronic inflammatory cell infiltrate.
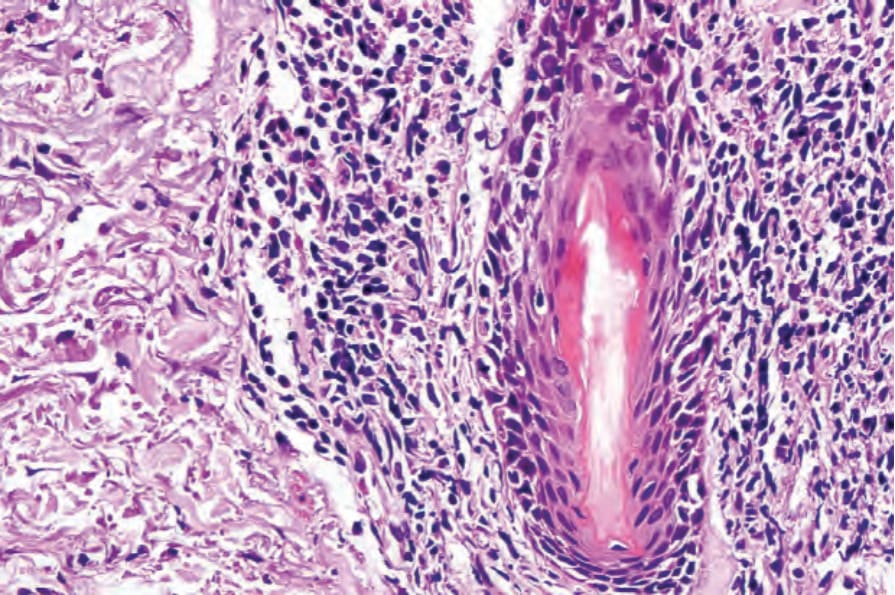
圖 17-48:盤狀紅斑性狼瘡——注意毛囊周圍的淋巴球浸潤。
Fig. 17.48 Discoid lupus erythematosus: note the perifollicular lymphocytic infiltrate.
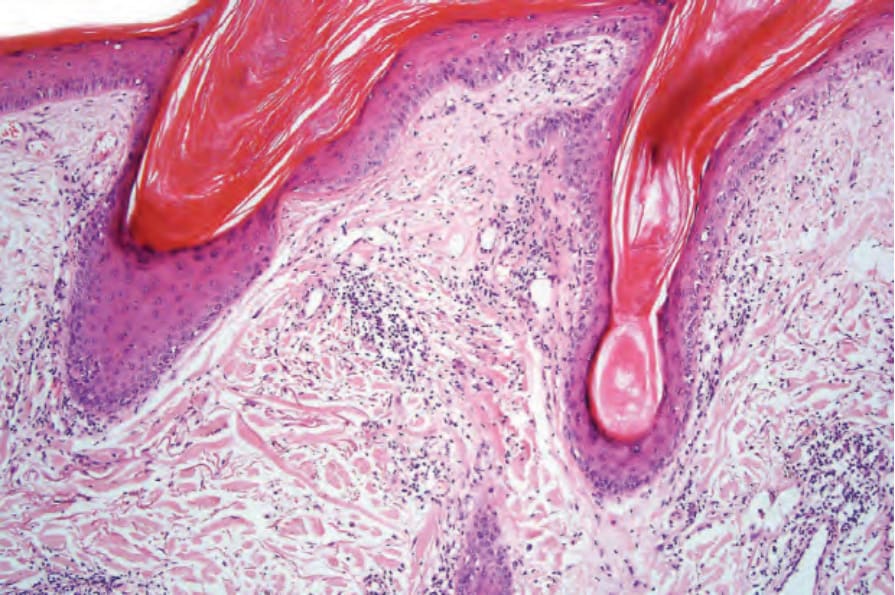
圖 17-49:盤狀紅斑性狼瘡——有明顯的毛囊栓塞。
Fig. 17.49 Discoid lupus erythematosus: there is marked follicular plugging.
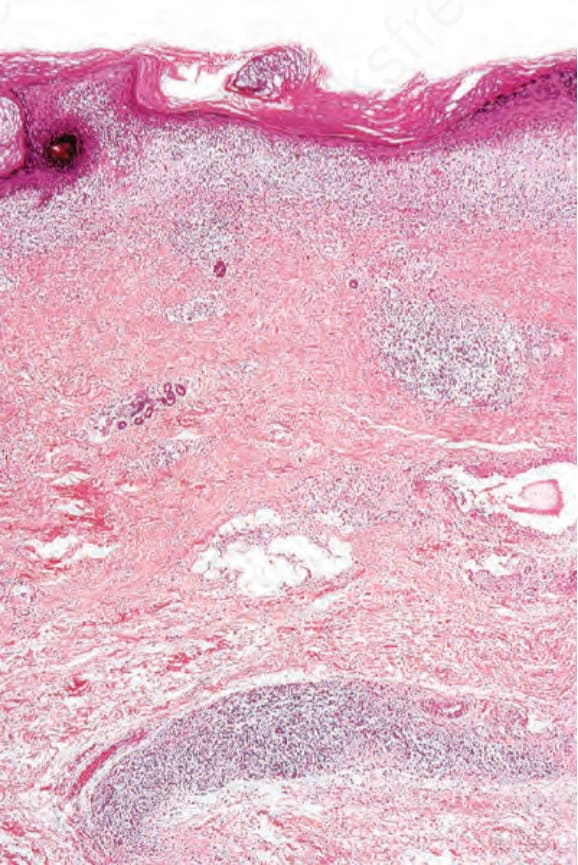
圖 17-50:盤狀紅斑性狼瘡——有過度角化與表皮萎縮。
Fig. 17.50 Discoid lupus erythematosus: there is hyperkeratosis and atrophy of the epidermis.
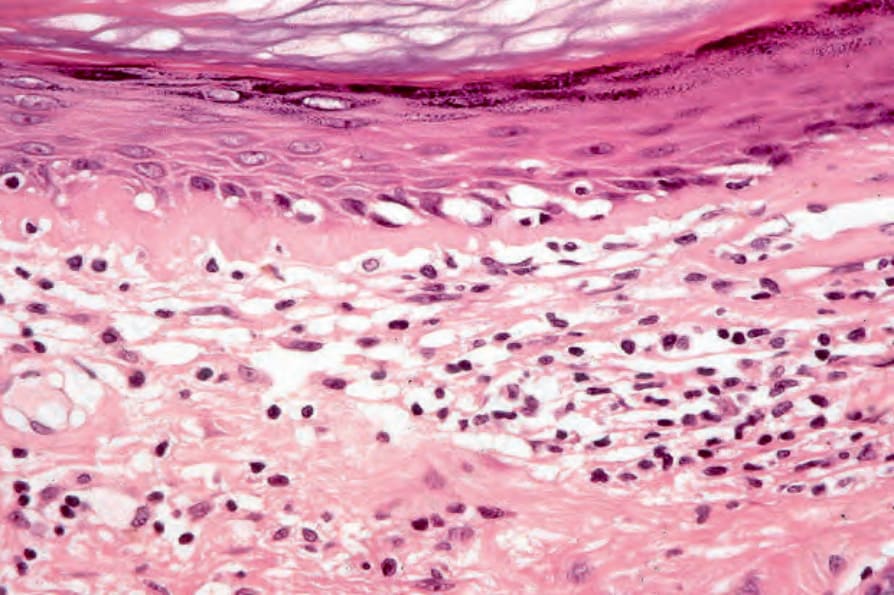
圖 17-51:盤狀紅斑性狼瘡——此視野中有過度角化、表皮萎縮與基底細胞水樣變性 (hydropic degeneration)。
Fig. 17.51 Discoid lupus erythematosus: in this view there is hyperkeratosis, epidermal atrophy, and basal cell hydropic degeneration.
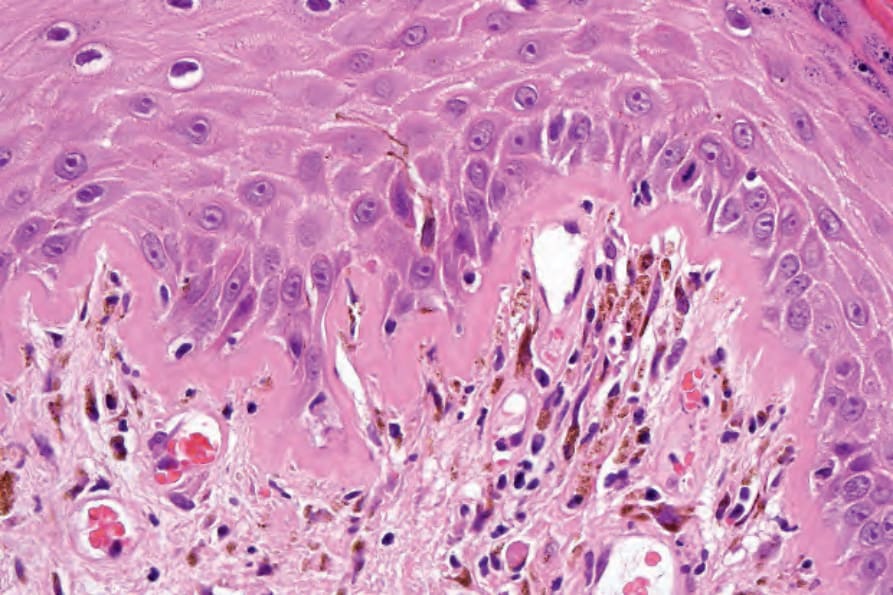
圖 17-52:盤狀紅斑性狼瘡——注意基底膜的非常明顯增厚與色素失禁。
Fig. 17.52 Discoid lupus erythematosus: note the very marked thickening of the basement membrane and pigmentary incontinence.
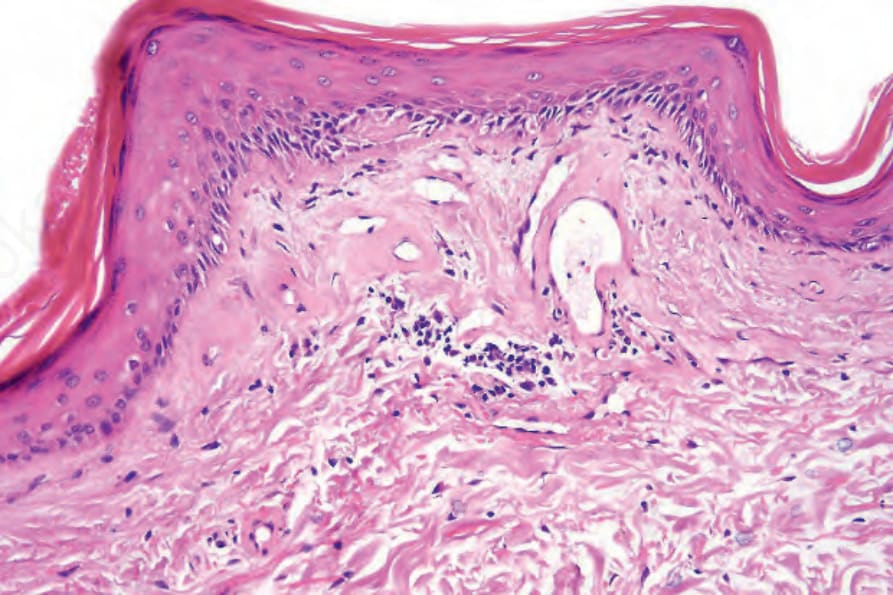
圖 17-53:盤狀紅斑性狼瘡——有明顯的血管壁增厚並伴玻璃樣變性 (hyalinization)。
Fig. 17.53 Discoid lupus erythematosus: there is marked blood vessel wall thickening with hyalinization.
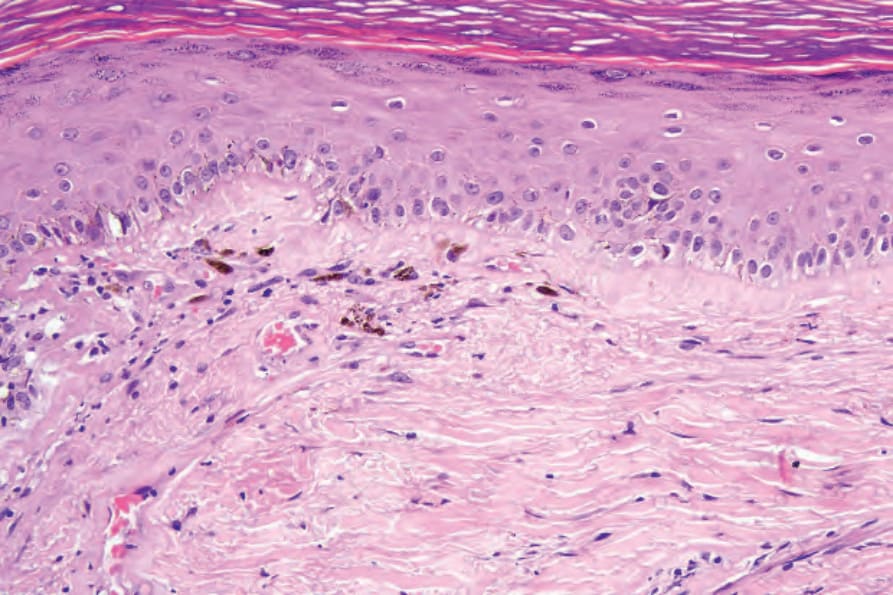
圖 17-54:盤狀紅斑性狼瘡——注意色素失禁。
Fig. 17.54 Discoid lupus erythematosus: note the pigmentary incontinence.
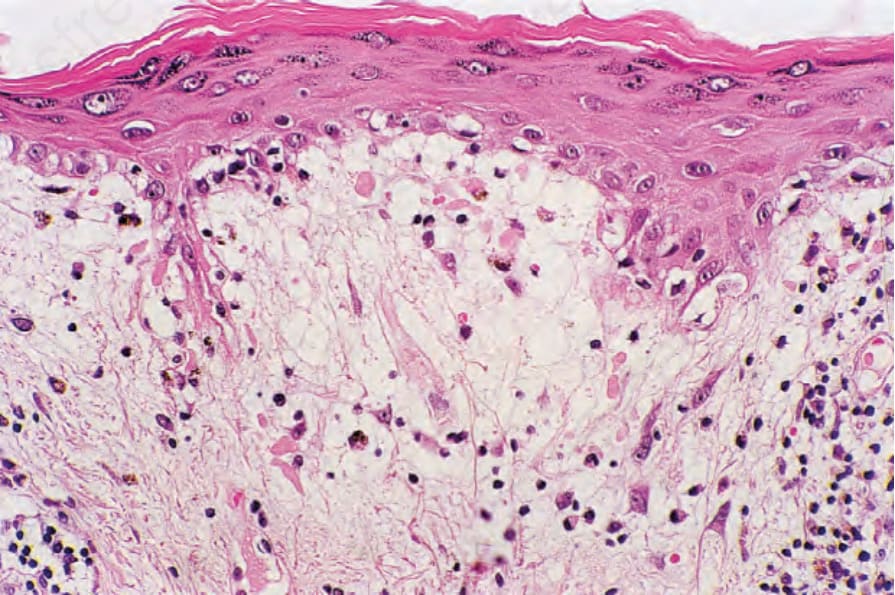
圖 17-55:盤狀紅斑性狼瘡——真皮乳頭中有數個類膠樣體 (cytoid bodies)。注意水樣變性。
Fig. 17.55 Discoid lupus erythematosus: several cytoid bodies are present in the papillary dermis. Note the hydropic degeneration.
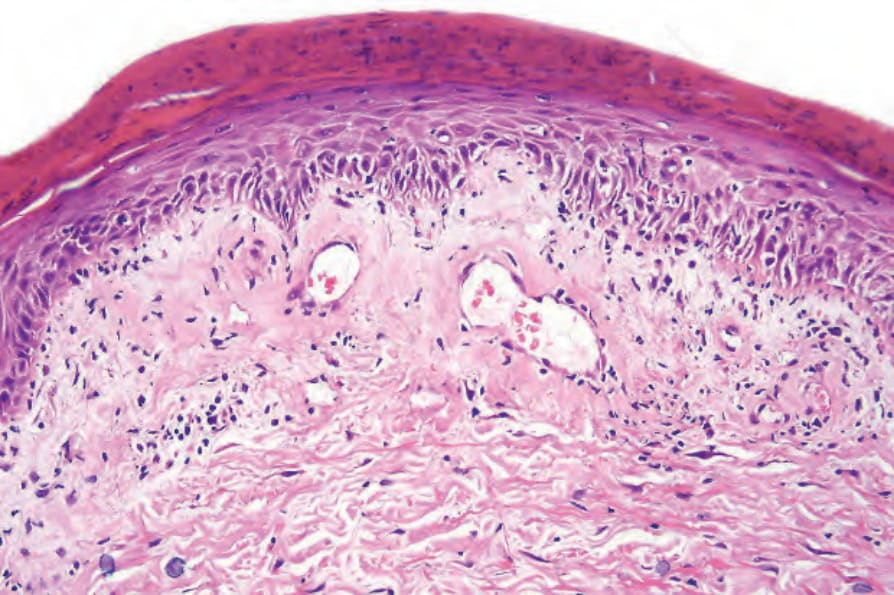
圖 17-56:盤狀紅斑性狼瘡——注意淺層真皮中的毛細血管擴張性血管。
Fig. 17.56 Discoid lupus erythematosus: note the telangiectatic vessels in the superficial dermis.
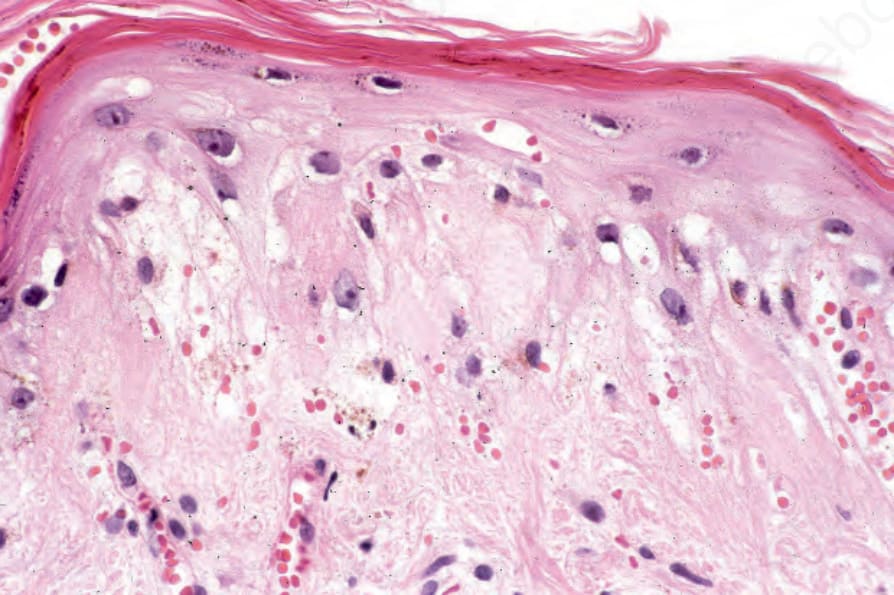
圖 17-57:盤狀紅斑性狼瘡——除了過度角化與表皮萎縮外,還有相當明顯的紅血球外滲。
Fig. 17.57 Discoid lupus erythematosus: in addition to hyperkeratosis and epidermal atrophy there is quite marked red cell extravasation.
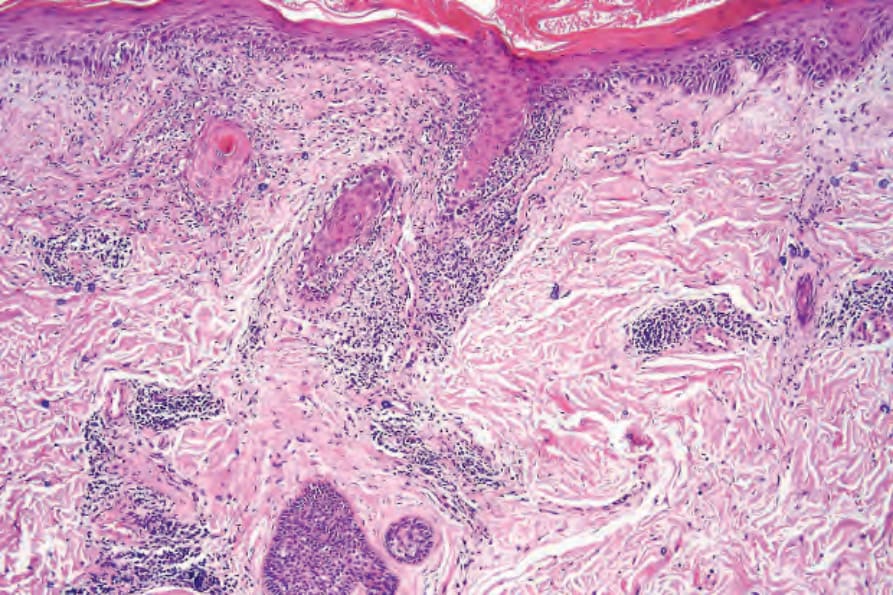
圖 17-58:盤狀紅斑性狼瘡——血管與毛囊被特徵性的濃厚淋巴組織細胞浸潤所包繞。
Fig. 17.58 Discoid lupus erythematosus: blood vessels and hair follicles are surrounded by a characteristic heavy lymphohistiocytic infiltrate.
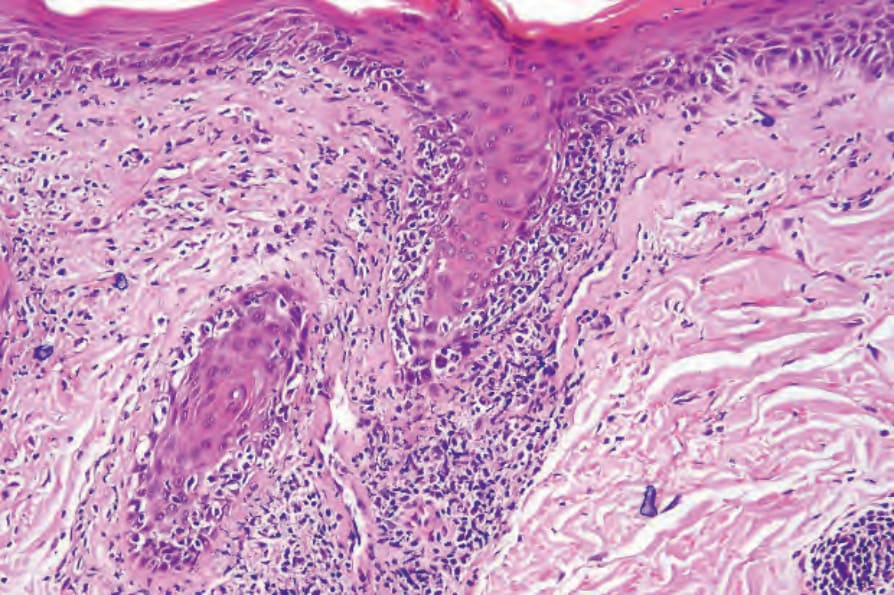
圖 17-59:盤狀紅斑性狼瘡——毛囊上皮顯示基底細胞水樣變性,並有濃厚的淋巴球浸潤。
Fig. 17.59 Discoid lupus erythematosus: the follicular epithelium shows basal cell hydropic degeneration and there is a heavy lymphocytic infiltrate.
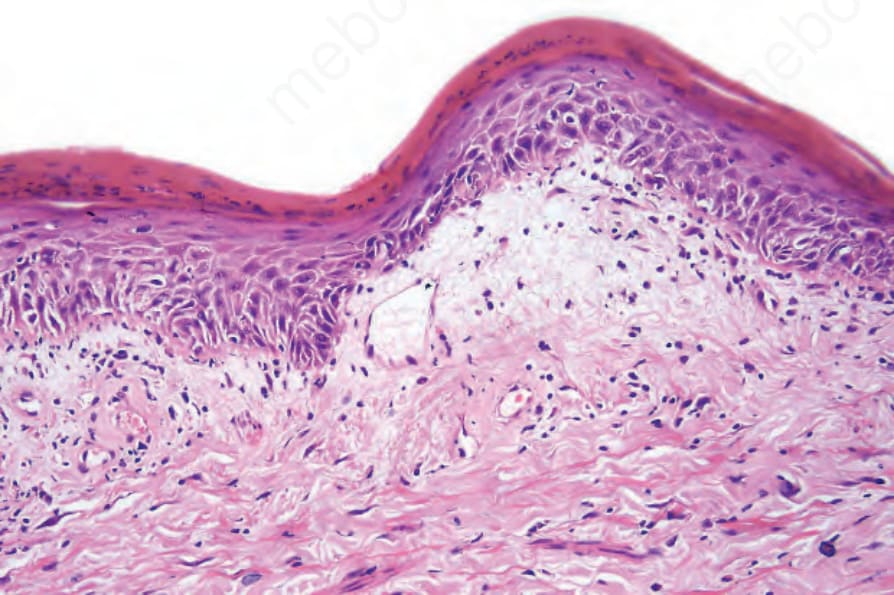
圖 17-60:盤狀紅斑性狼瘡——如此視野所示的基質黏液沉積並不少見。
Fig. 17.60 Discoid lupus erythematosus: stromal mucin deposition, as seen in this field, is not uncommon.
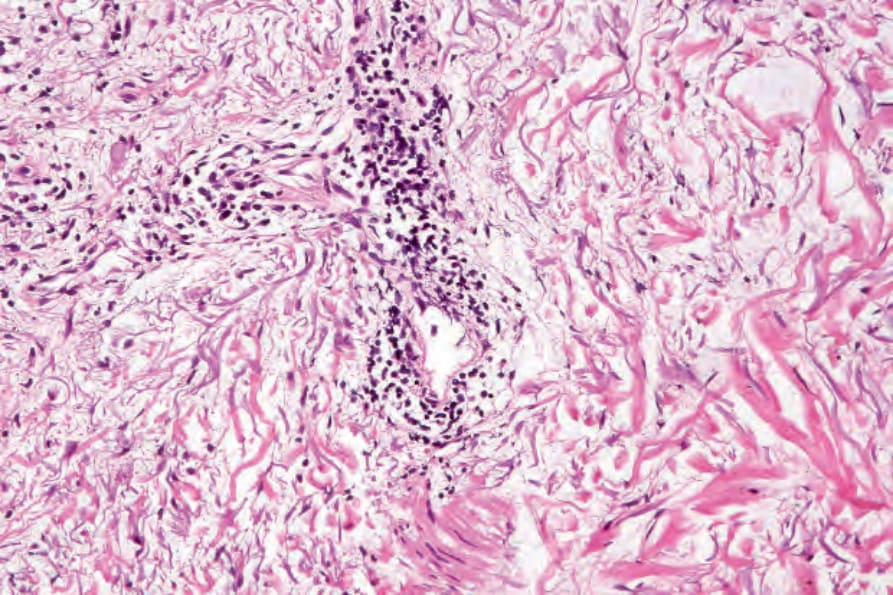
圖 17-61:腫脹性盤狀紅斑性狼瘡——在此罕見變異型中,有非常明顯的黏液沉積。
Fig. 17.61 Tumid discoid lupus erythematosus: in this rare variant, there is very marked mucin deposition. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, USA.
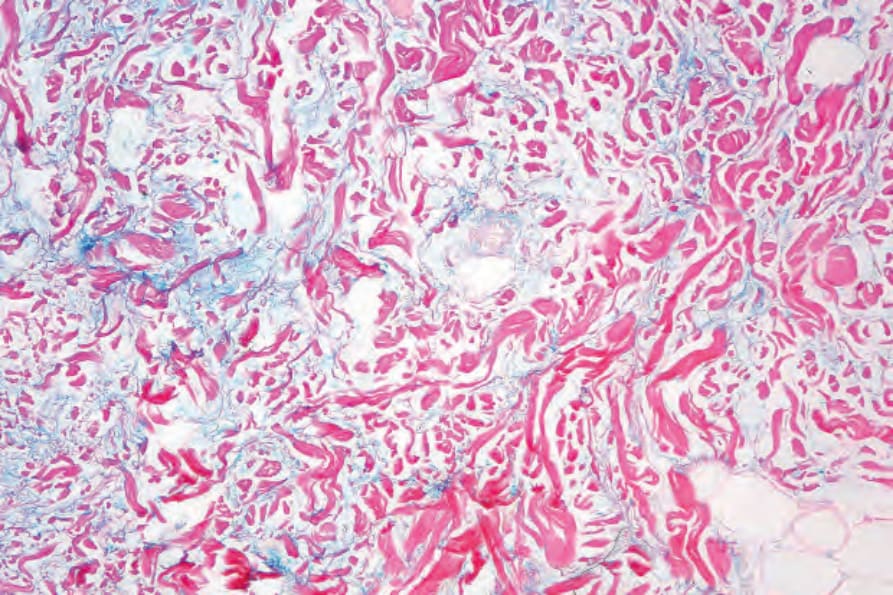
圖 17-62:腫脹性盤狀紅斑性狼瘡——黏液以 Alcian blue(pH 2.5)染色。
Fig. 17.62 Tumid discoid lupus erythematosus: the mucin stains with Alcian blue, pH 2.5. By courtesy of J. Cohen, MD, Dermatopathology Laboratory, Tucson, USA.
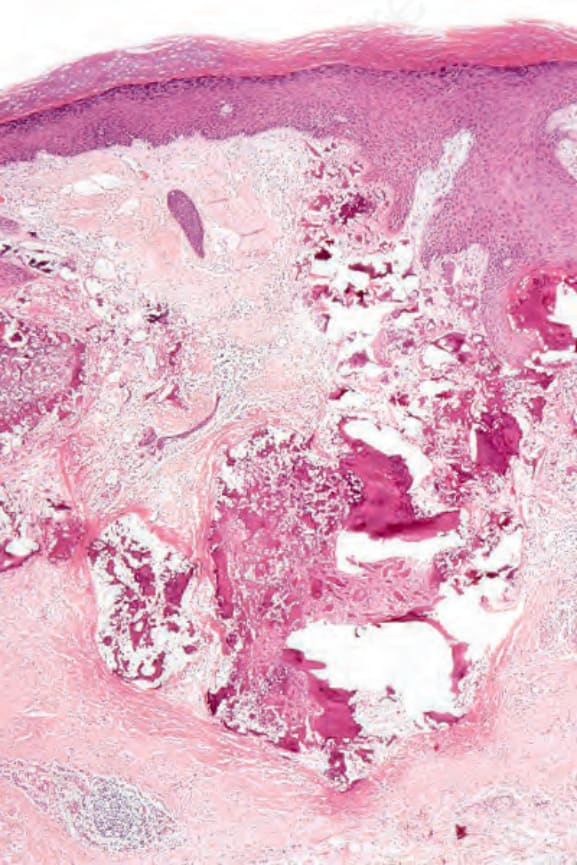
圖 17-63:盤狀紅斑性狼瘡——鈣化是罕見的特徵。此例中有顯著的經表皮排出 (transepidermal elimination)。
Fig. 17.63 Discoid lupus erythematosus: calcification is a rare feature. In this example, there is striking transepidermal elimination.
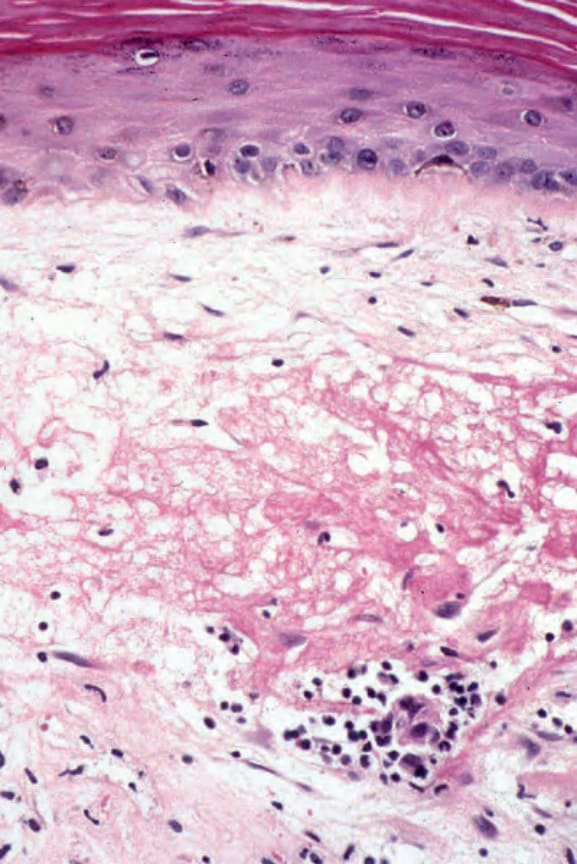
圖 17-64:盤狀紅斑性狼瘡——注意類纖維蛋白壞死 (fibrinoid necrosis) 的存在。這更常是全身性變異型的特徵。
Fig. 17.64 Discoid lupus erythematosus: note the presence of fibrinoid necrosis. This is more usually a feature of the systemic variant.
在 DLE 的癒合病灶中有過度角化,表皮可能萎縮,或更常見地,略為增厚。基底膜區域特徵性地明顯增厚。真皮纖維化,有時程度可達類似硬化性苔癬 (lichen sclerosus) (Fig. 17.65)。在有毛部位,特別是頭皮,可能有非常明顯的毛囊栓塞並伴隨慢性發炎,且在進展期疾病中常有毛皮脂腺結構的完全喪失,並被膠原纖維束所取代 (Fig. 17.66)。皮脂腺萎縮或喪失在頭皮侵犯中早期發生,而慢性發炎變化集中於毛囊中段的層級。有人提出此處可能代表毛囊幹細胞的灶區,因此此部位的慢性發炎很容易造成永久性掉髮。源自退化表皮角質形成細胞的繼發性類澱粉沉積,偶爾可在真皮乳頭中偵測到。
罕見情況下,DLE 可能表現為濃密的淺層與深層血管周圍及附屬器周圍浸潤,而無顯著的表皮變化:因此在組織學上無法與 Jessner 淋巴球浸潤 (lymphocytic infiltrate of Jessner) 及多形性日光疹區分。
在肥厚型病灶中,除了基底細胞損傷的特徵外,還有明顯的過度角化、顆粒層增厚 (hypergranulosis) 與不規則棘層肥厚並伴乳頭瘤狀增生 (papillomatosis)。類膠樣體在下表皮常很明顯。曾報告類澱粉沉積為常見的發現。其特徵在組織學上常無法與肥厚型扁平苔癬區分。曾在肥厚型紅斑性狼瘡中報告表皮內彈性纖維 (intraepidermal elastic fibers) 的存在,常伴隨彈性變性物質的經表皮排出。這不是扁平苔癬的特徵。有時可模擬消退中或演變中的角化棘皮瘤。
來自狼瘡凍瘡 (lupus pernio) 病人的切片顯示袖套狀的血管周圍淋巴球浸潤並伴水腫、紅血球外滲與不等的血管類纖維蛋白變化 (Figs 17.67–17.70)。部分切片顯示明顯的淋巴球性血管炎 (lymphocytic vasculitis)。發炎浸潤常延伸至深層真皮與皮下脂肪組織。交界面表皮變化,範圍從局部空泡變化到苔癬樣組織反應 (lichenoid tissue reaction),常存在。有時可見汗腺周圍的慢性發炎。
黏膜病灶常難以與扁平苔癬區分。它們以過度角化為特徵,常伴隨角化不全。上皮可能萎縮或棘層肥厚。基底角質形成細胞的水樣變性,有時伴隨類膠樣體形成,伴隨濃密的淋巴組織細胞浸潤,其中漿細胞常很多。深層血管周圍慢性發炎細胞浸潤的存在傾向支持 DLE 的診斷。然而,常需直接免疫螢光來建立診斷。
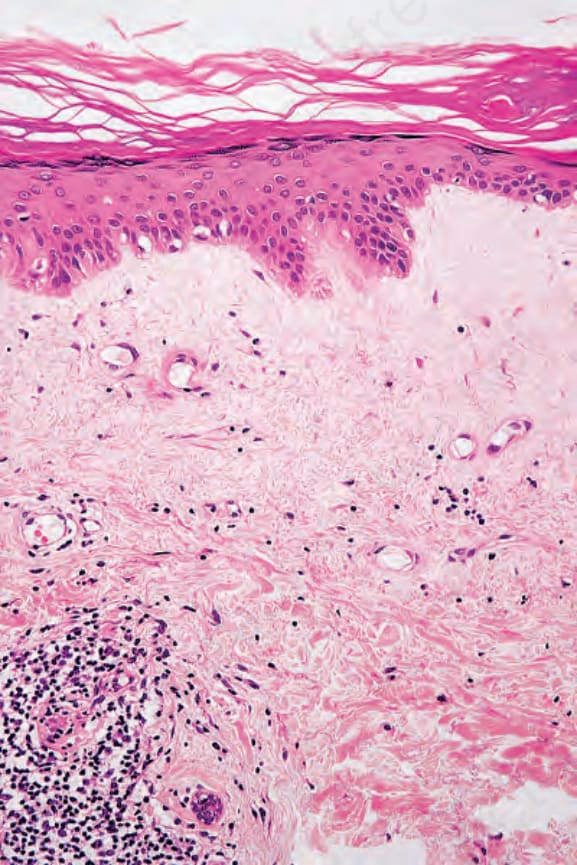
圖 17-65:盤狀紅斑性狼瘡——此例中,淺層真皮硬化的存在令人聯想到硬化性苔癬 (lichen sclerosus)。
Fig. 17.65 Discoid lupus erythematosus: in this example, the presence of superficial dermal sclerosis is reminiscent of lichen sclerosus.
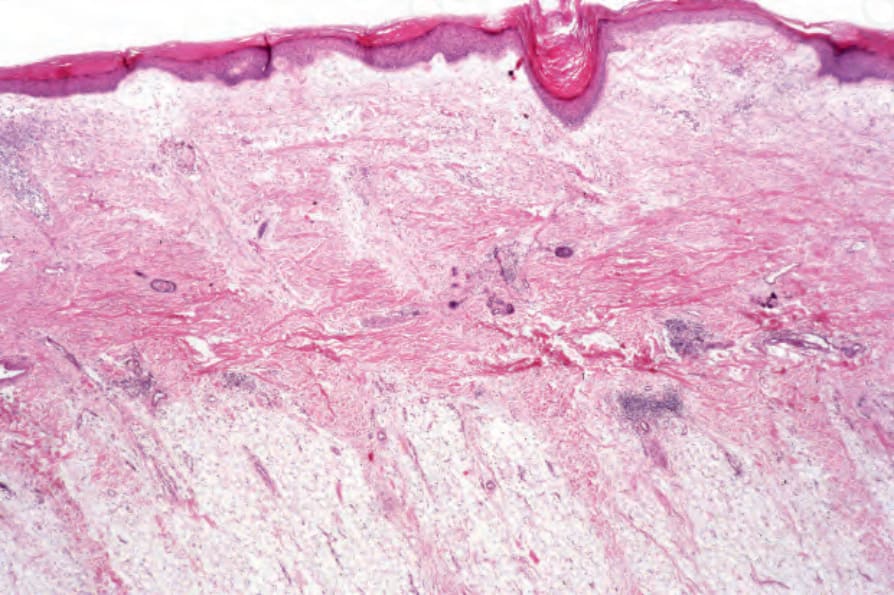
圖 17-66:盤狀紅斑性狼瘡——注意此頭皮標本中毛囊的完全缺失。
Fig. 17.66 Discoid lupus erythematosus: note the complete absence of hair follicles in this scalp specimen.
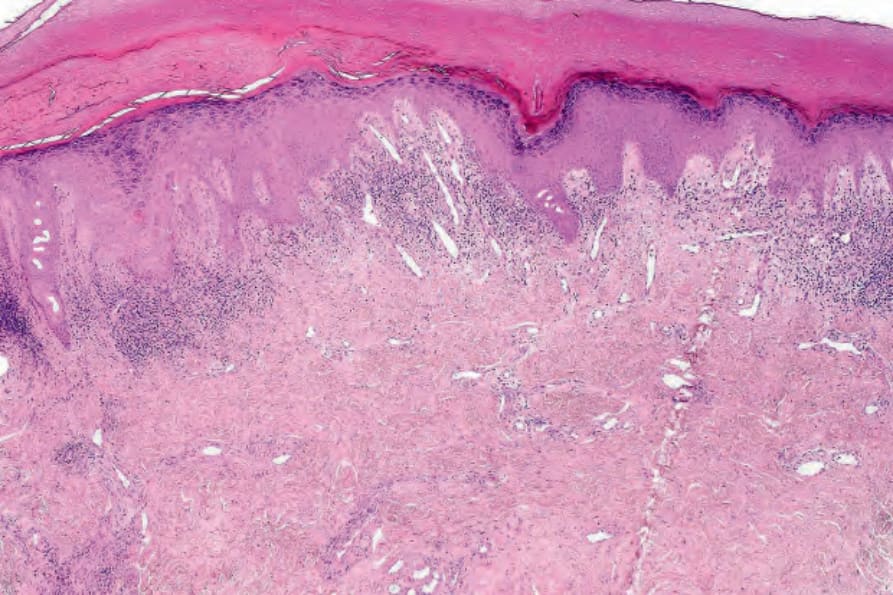
圖 17-67:凍瘡型紅斑性狼瘡——有不規則棘層肥厚與淺層淋巴球浸潤。擴張的血管 (ectatic vessels) 很明顯。
Fig. 17.67 Chilblain lupus erythematosus: there is irregular acanthosis and a superficial lymphocytic infiltrate. Ectatic vessels are conspicuous.
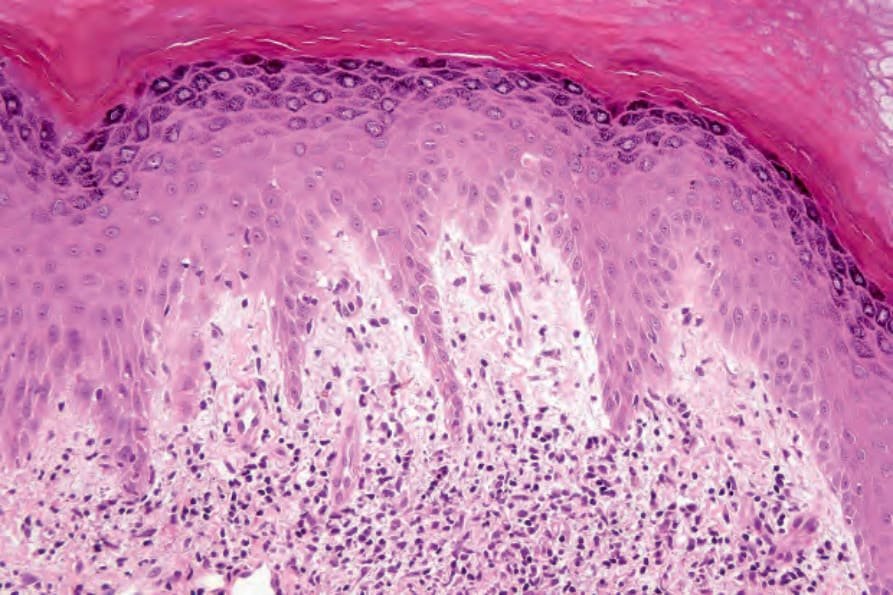
圖 17-68:凍瘡型紅斑性狼瘡——此視野中有真皮乳頭的明顯水腫。
Fig. 17.68 Chilblain lupus erythematosus: in this field, there is marked edema of the papillary dermis.
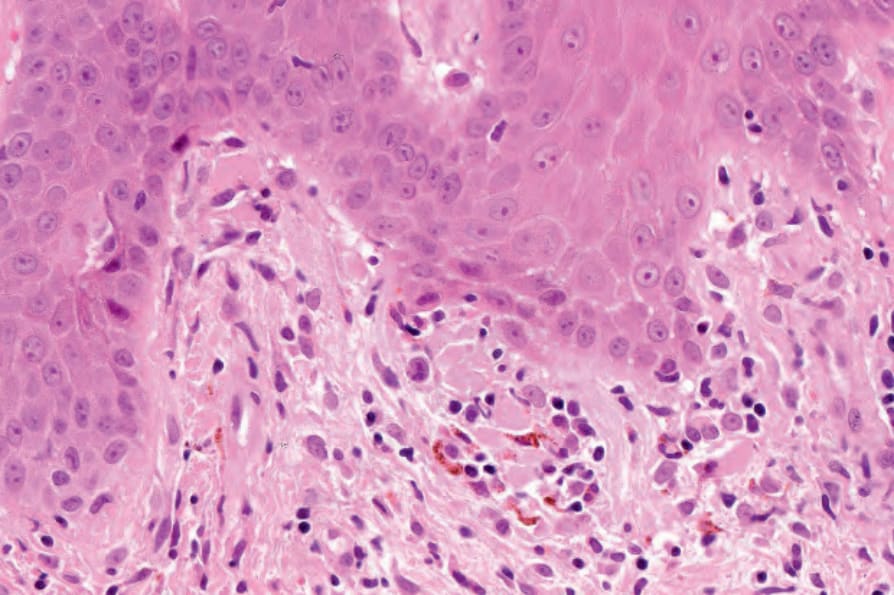
圖 17-69:凍瘡型紅斑性狼瘡——有局部基底細胞水樣變性,類膠樣體 (cytoid bodies) 很明顯。
Fig. 17.69 Chilblain lupus erythematosus: there is focal basal cell hydropic degeneration, and cytoid bodies are conspicuous.
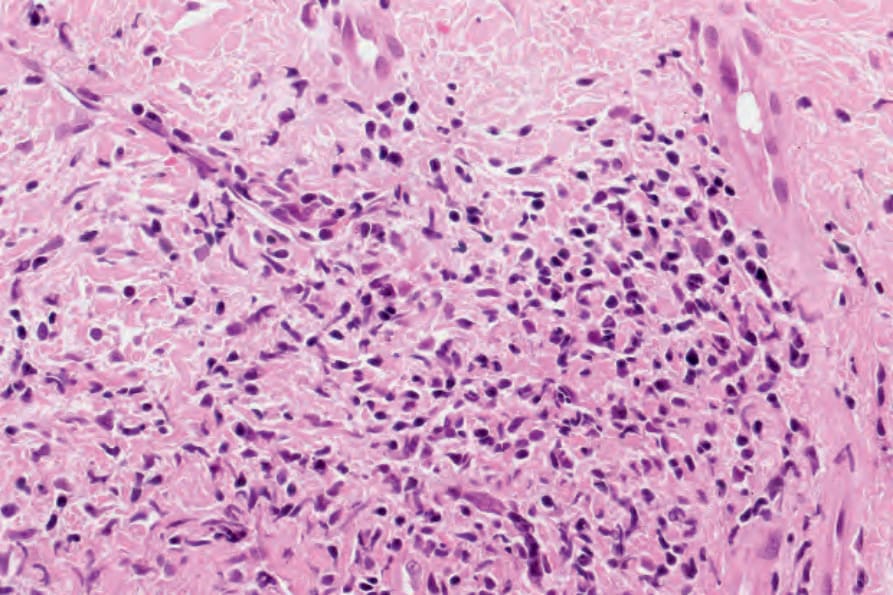
圖 17-70:凍瘡型紅斑性狼瘡——有濃密的淋巴球浸潤。
Fig. 17.70 Chilblain lupus erythematosus: there is a dense lymphocytic infiltrate.
深部紅斑性狼瘡 (Lupus erythematosus profundus)
深部紅斑性狼瘡的組織學特徵於第 10 章中討論。
亞急性皮膚紅斑性狼瘡 (Subacute cutaneous lupus erythematosus)
雖然 SCLE 的組織學特徵與 DLE 的特徵顯示相當大的重疊,但仍有診斷指標。在 SCLE 中,過度角化傾向輕微,萎縮較明顯,且表皮脊型態常消失 (Fig. 17.71)。有時角化不全可能是一個特徵。基底膜增厚極微或缺如,毛囊常不受影響或僅顯示輕微的角質栓塞。在 DLE 中,發炎細胞浸潤較濃密,占據真皮乳頭與網狀真皮,且常顯示毛囊周圍的強調。在 SCLE 中,淋巴球外移 (lymphocytic exocytosis) 可能很明顯,衛星細胞壞死 (satellite cell necrosis) 並不少見 (Fig. 17.72)。表皮基底層的液化變性通常存在,雖然常僅為輕微 (Figs 17.73 and 17.74)。然而有時它足夠明顯,以致造成表皮下水疱形成。偶爾可見真皮乳頭膠原蛋白的均質化。膠樣體 (colloid body) 形成與色素失禁常不明顯,但有時是主要特徵。發炎細胞浸潤典型上輕微、位於淺層、且呈血管周圍分布。然而在某些例子中,它表現為苔癬樣帶。真皮發炎細胞浸潤中也可見不等量的嗜伊紅性球。這是一個完全非特異性的發現,與可能的藥物誘發性 SCLE 無關。
無 SCLE 且具 SS-A (Ro) 抗體之病人,其皮膚病灶的組織病理特徵與 SCLE 病灶所見者非常相似。這些病人常有不同的臨床疾病,包括 Sjögren 症候群與類風濕性關節炎。
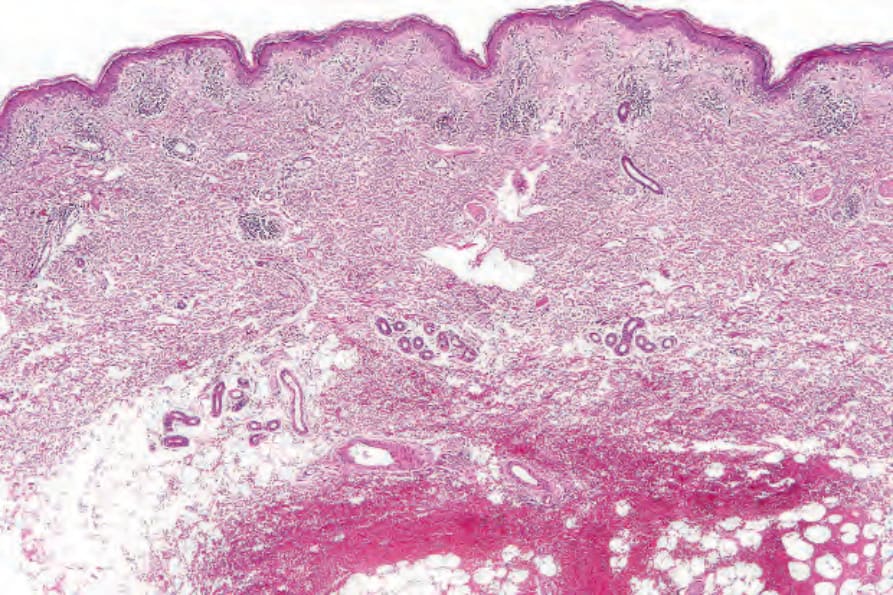
圖 17-71:亞急性皮膚紅斑性狼瘡——低倍視野顯示輕微過度角化。有血管周圍的慢性發炎細胞浸潤。
Fig. 17.71 Subacute cutaneous lupus erythematosus: low-power view showing slight hyperkeratosis. A perivascular chronic inflammatory cell infiltrate is present.
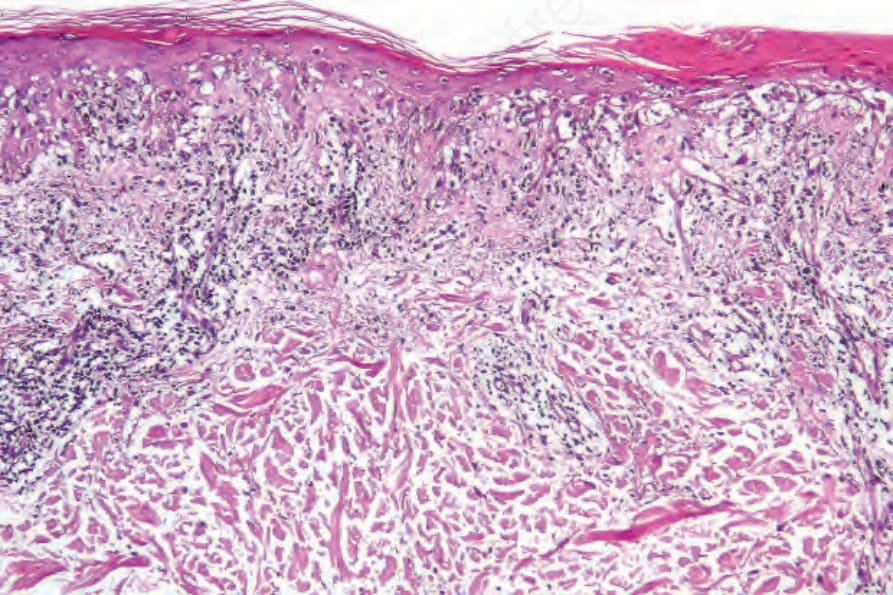
圖 17-72:亞急性皮膚紅斑性狼瘡——此例中,除了基底細胞水樣變性外,還有表皮細胞間水腫並伴淋巴球外移。
Fig. 17.72 Subacute cutaneous lupus erythematosus: in this example, in addition to basal cell hydropic degeneration, there is intercellular edema of the epidermis with lymphocytic exocytosis.
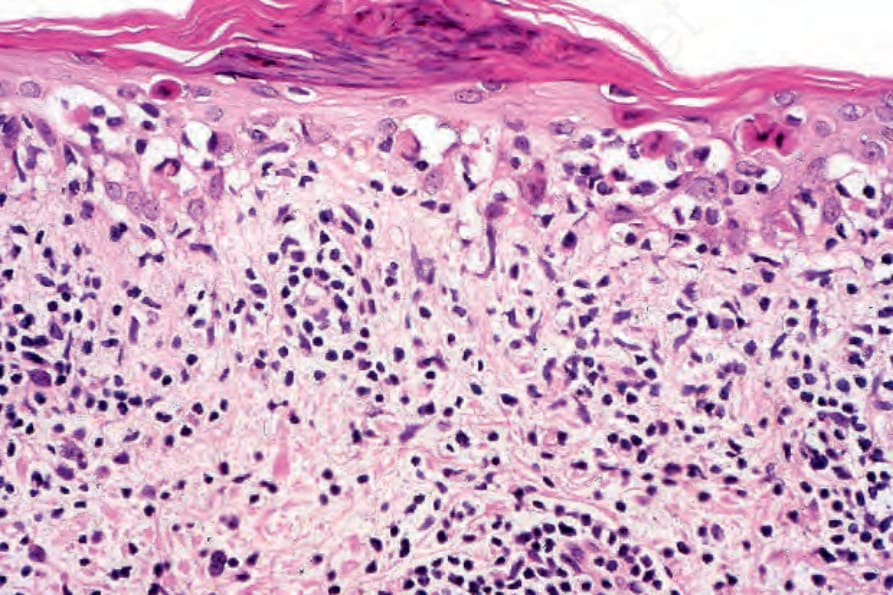
圖 17-73:亞急性皮膚紅斑性狼瘡——注意眾多類膠樣體與局部衛星細胞壞死的存在。
Fig. 17.73 Subacute cutaneous lupus erythematosus: note the presence of numerous cytoid bodies and focal satellite cell necrosis.
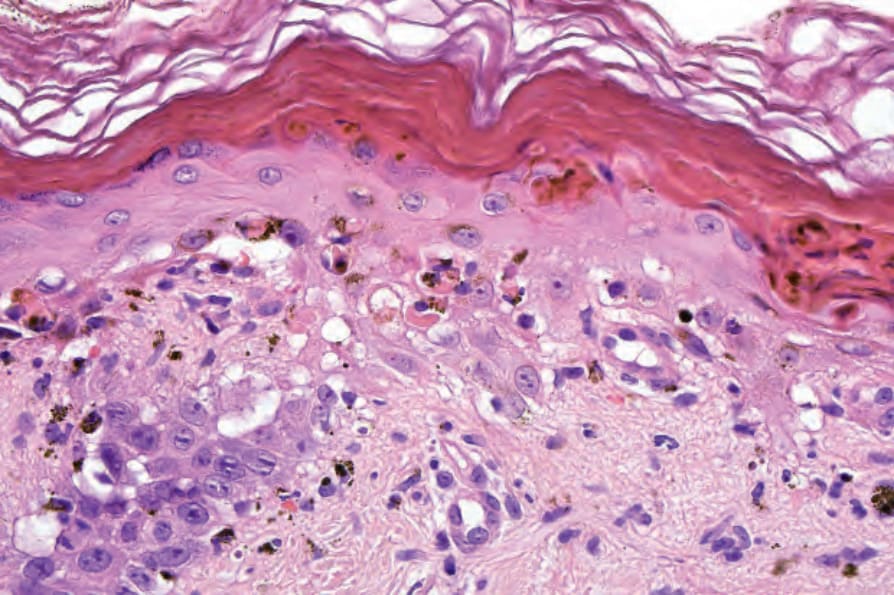
圖 17-74:亞急性皮膚紅斑性狼瘡——此例中有明顯的基底細胞水樣變性與細胞凋亡。僅存在斑塊狀的慢性發炎細胞浸潤。
Fig. 17.74 Subacute cutaneous lupus erythematosus: in this example, there is marked basal cell hydropic degeneration and apoptosis. Only a patchy chronic inflammatory cell infiltrate is present.
全身性紅斑性狼瘡之嗜中性球皮膚病 (Neutrophilic dermatosis of the systemic lupus erythematosus)
SLE 嗜中性球皮膚病的組織學特徵多變。嗜中性球浸潤的強度典型上從真皮乳頭中少細胞的嗜中性球浸潤,到血管周圍與間質分布中度至高度細胞性的嗜中性球浸潤不等,常混雜有橫跨整個真皮的核碎屑 (nuclear debris),可能延伸至皮下脂肪組織。嗜中性球發炎細胞浸潤一般伴隨白血球碎裂,而血管炎按慣例缺如。除了真皮嗜中性球浸潤外,最近一篇論文發現,表皮、毛囊、皮脂腺與汗腺之上皮內嗜中性球的存在,高度提示嗜中性球蕁麻疹性皮膚病。其他組織學特徵,作為可能與紅斑性狼瘡關聯之有用指標,以不等的程度與頻率存在,包括交界面皮膚炎 (interface dermatitis)、基底角質形成細胞的空泡變性、基底膜增厚、毛囊栓塞與真皮黏液沉積。直接免疫螢光檢測通常顯示陽性狼瘡帶試驗。
組織學鑑別診斷主要取決於嗜中性球浸潤的強度。真皮乳頭中少細胞嗜中性球浸潤的例子,主要應與大疱型 SLE、疱疹樣皮膚炎與線狀 IgA 病 (linear IgA disease) 區分——免疫螢光分析對此區分至關重要。較顯著嗜中性球浸潤的鑑別診斷包括史迪爾病、Sweet 症候群、貝賽特病 (Behçet disease) 與感染。
全身性紅斑性狼瘡 (Systemic lupus erythematosus)
SLE 的組織學特徵多變。早期病灶的變化可能非常輕微而隱微,常僅包含略微的表皮基底細胞液化變性、真皮乳頭水腫,以及輕度的慢性發炎細胞浸潤,例如見於顴部紅斑 (Fig. 17.75)。在其他情況下,外觀類似 SCLE。
SLE 的組織學常無法與盤狀變異型區分 (Fig. 17.76)。然而,真皮膠原蛋白的類纖維蛋白變性 (fibrinoid degeneration) 在 SLE 中比在 DLE 中更常見。膠原束之間常可見真皮黏液沉積。在多數案例中,此特徵很隱微,但黏液沉積可能非常顯著。增厚的基底膜在 DLE 病灶中更常見。如同 DLE,最常見的細胞類型是 T 淋巴球,但哪個亞群占多數尚不確定。CD4+ 與 CD4+ 亞群占優勢皆曾被記載。這可能是因為病灶年齡、治療效應與抗體專一性的差異可解釋此明顯的異常。CD4+ 淋巴球在基底角質形成細胞水樣變性的部位大量存在於表皮中。
抗磷脂症候群皮膚病灶的組織病理特徵包含靜脈與動脈血栓,而無任何血管炎的證據 (Figs 17.77–17.79)。在早期病灶中,內皮細胞損傷可能明顯,紅血球外滲常見。較舊的病灶以明顯的血管增生為特徵,常呈小葉狀分布並伴隨含鐵血黃素沉積 (hemosiderosis)。有時可見鞋釘狀 (hobnail) 反應性內皮細胞與嗜伊紅性透明小球 (eosinophilic hyaline globules)。常可見伴隨不等數量漿細胞的淋巴球浸潤。在某些病人中,曾記載 Degos 病與皮膚鬆弛症 (anetoderma) 的臨床與組織學特徵。
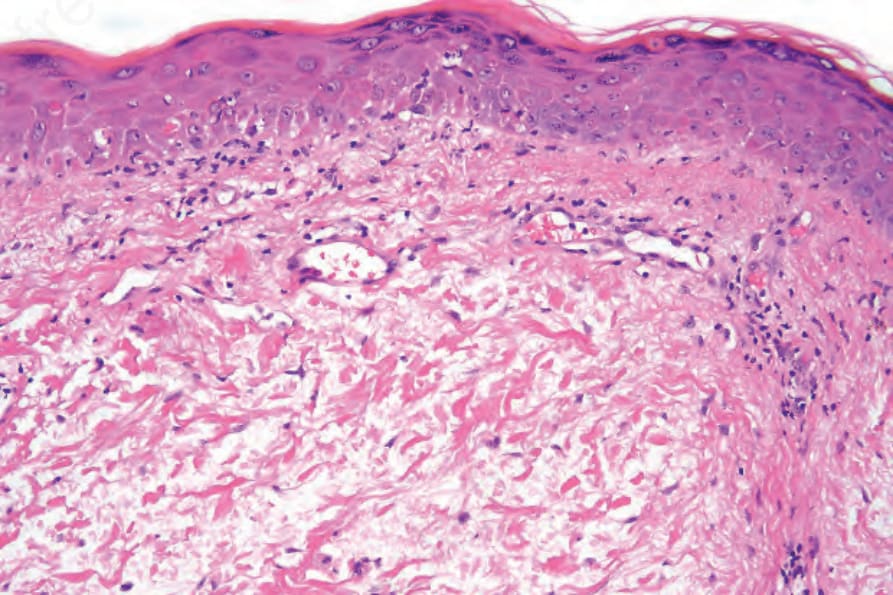
圖 17-75:全身性紅斑性狼瘡——雖然變化可能與盤狀或亞急性變異型相同,但有時特徵很輕微,如此例。有局部基底細胞水樣變性、細胞凋亡與毛細血管擴張。
Fig. 17.75 Systemic lupus erythematosus: although the changes may be identical to the discoid or subacute variants, sometimes the features are mild, as in this case. There is focal basal cell hydropic degeneration, apoptosis, and telangiectasia.
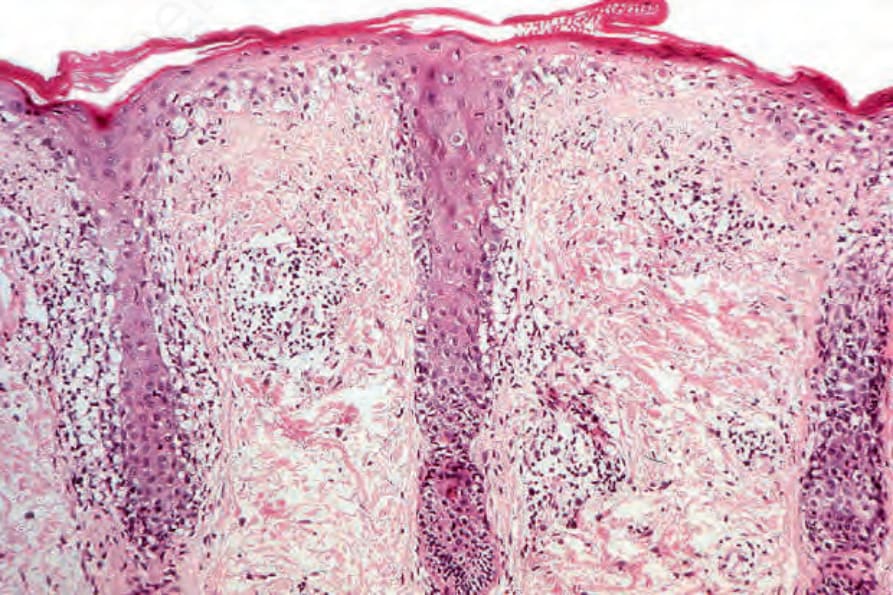
圖 17-76:全身性紅斑性狼瘡——此例中,特徵類似盤狀變異型。注意過度角化、表皮萎縮、毛囊侵犯與嚴重的基底細胞水樣變性。有血管周圍與毛囊周圍的慢性發炎細胞浸潤。
Fig. 17.76 Systemic lupus erythematosus: in this example, the features resemble the discoid variant. Note the hyperkeratosis, epidermal atrophy, follicular involvement, and gross basal cell hydropic degeneration. A perivascular and perifollicular chronic inflammatory cell infiltrate is present.
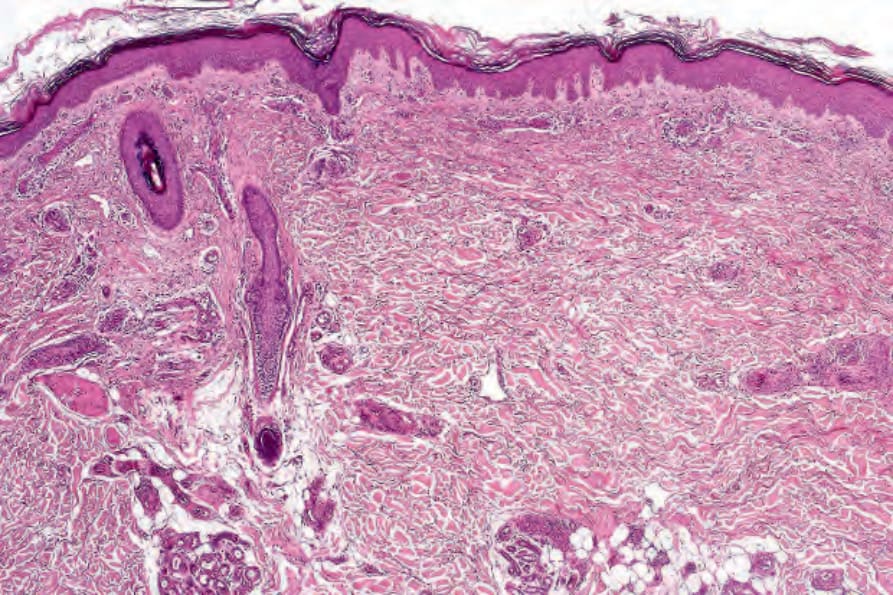
圖 17-77:狼瘡抗凝血劑症候群——低倍視野顯示淺層血管被小血栓阻塞。
Fig. 17.77 Lupus anticoagulant syndrome: low-power view showing superficial blood vessels occluded by small thrombi.
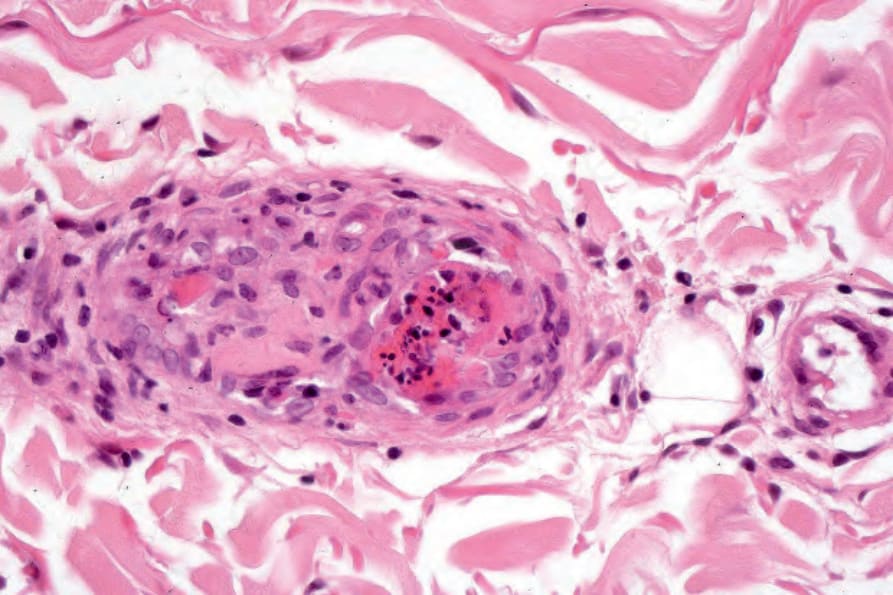
圖 17-78:狼瘡抗凝血劑症候群——阻塞血管的特寫——注意核碎屑 (nuclear debris)。
Fig. 17.78 Lupus anticoagulant syndrome: close-up view of occluded vessel – note the nuclear debris.
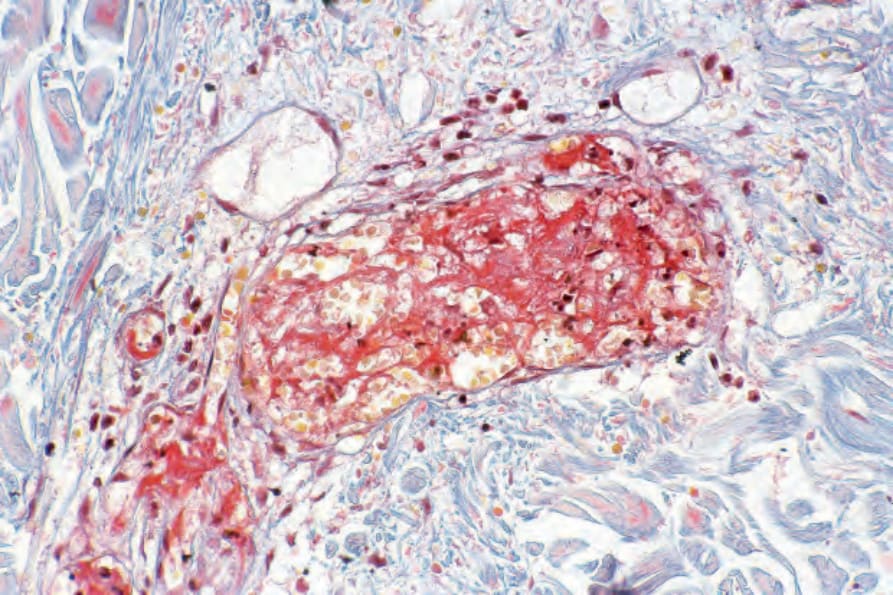
圖 17-79:狼瘡抗凝血劑症候群——在此 phosphotungstic acid–hematoxylin 染色切片中確認纖維蛋白 (fibrin) 的存在。
Fig. 17.79 Lupus anticoagulant syndrome: the presence of fibrin is confirmed in this phosphotungstic acid–hematoxylin stained section.
腎臟病灶 (Renal lesions)
腎臟病灶可在 70% 至 80% 的病人中偵測到。組織學特徵依據國際腎臟病學會/腎臟病理學會 (International Society of Nephrology/Renal Pathology Society) 2003 年狼瘡腎炎分類細分為六類,代表先前 WHO 分類的演進:
- 在第 I 類病灶(極微繫膜性狼瘡腎炎 minimal mesangial lupus nephritis)中,光學顯微鏡無法偵測到異常。免疫複合體的繫膜沉積可藉由免疫螢光、電子顯微鏡或兩者鑑定。
- 第 II 類病灶(繫膜增生性狼瘡腎炎 mesangial proliferative lupus nephritis)以任何程度的繫膜細胞增多(定義為 3 微米厚切片中每個繫膜區域有三個或以上的繫膜細胞)並伴繫膜免疫沉積為特徵。偶爾可藉由免疫螢光或電子顯微鏡偵測到少量涉及周邊毛細血管壁的小型免疫沉積。不可接受的特徵為光學顯微鏡下的內皮下沉積,以及任何由先前腎絲球內皮細胞增生、壞死或新月體所造成的節段性或全面性腎絲球瘢痕。免疫螢光顯示 IgG 與補體的繫膜沉積,電子顯微鏡可偵測到繫膜電子緻密沉積物。繫膜性狼瘡腎絲球腎炎是腎絲球病灶最輕微的型態,存在於約 10% 有腎臟侵犯的病人。
- 第 III 類病灶(局部狼瘡腎炎 focal lupus nephritis)以活動性或非活動性的局部、節段性或全面性的內毛細血管或外毛細血管腎絲球腎炎為區別特徵,侵犯所有腎絲球的 < 50%,伴隨局部內皮下免疫沉積,伴或不伴繫膜增生。局部節段性增生性腎絲球腎炎見於約 30% 的病人。在此變異型中,只有散在的腎絲球受影響(局部),且通常只有腎絲球簇的一部分受侵犯(節段性)。病灶可能為增生性與壞死性。受侵犯的腎絲球通常顯示繫膜與內皮增生、多形核細胞浸潤、纖維蛋白沉積與核碎裂 (karyorrhexis);偶爾可見血鐵質小體 (hematoxylin bodies)。血鐵質小體被視為 SLE 的組織學標誌。它們是圓形至卵圓形的結構,以蘇木紫與伊紅染色呈紫/紅-粉紅/藍色。它們對 Feulgen 反應呈陽性染色,von Kossa 染色為陰性。血鐵質小體被認為是 LE 細胞現象的體內 (in vivo) 對應物。雖然常被尋找,但它們常很難找到。電子顯微鏡典型上顯示繫膜與內皮下電子緻密(免疫複合體)沉積物。臨床上,病人表現為血尿與蛋白尿,雖然一部分可能發展出慢性腎衰竭。
- 第 IV 類病灶(瀰漫性狼瘡腎炎 diffuse lupus nephritis)以活動性或非活動性的局部、節段性或全面性的內毛細血管或外毛細血管腎絲球腎炎為特徵,侵犯所有腎絲球的 ≥ 50%,伴隨瀰漫性內皮下免疫沉積,伴或不伴繫膜增生。它們可進一步細分為瀰漫性節段性 (IV-S) 與瀰漫性全面性 (IV-G) 狼瘡腎炎。瀰漫性狼瘡腎絲球腎炎發生於約 50% 有腎炎/腎病症候群或高血壓的 SLE 病人。幾乎所有腎絲球皆受影響,且除了繫膜與內皮增生外,上皮細胞也可能參與,形成新月體。仔細檢查腎絲球簇的周邊常顯示規則增厚的毛細血管——「線圈」(‘wire-loop’) 病灶,被認為對 SLE 具特異性。
- 第 V 類(狼瘡膜性腎絲球腎炎 lupus membranous glomerulonephritis)病灶顯示全面性或節段性的上皮下顆粒狀免疫沉積,伴或不伴繫膜改變。也可能存在散在的內皮下免疫沉積。第 V 類病灶可與第 III 類及第 IV 類病灶相關。狼瘡膜性腎絲球腎炎見於約 10% 的 SLE 病人,表現為蛋白尿或腎病症候群。組織學上,有腎絲球毛細血管的均勻增厚而無顯著的發炎細胞浸潤。可能存在進展期腎絲球硬化。在超微結構上,電子緻密沉積物見於上皮下。
- 第 VI 類病灶(進展期硬化性狼瘡腎炎 advanced sclerosis lupus nephritis)以 ≥ 90% 的腎絲球出現全面性硬化而無殘餘活動性為特徵,與狼瘡腎炎相關。它們可代表第 III、IV 或 V 類病灶的進展。除了腎絲球病灶外,有腎臟侵犯的病人可能呈現急性壞死性血管炎。
心臟、肺臟、淋巴結等器官病灶
心臟在約 50% 的 SLE 病人中受侵犯。類纖維蛋白變性與血鐵質小體的急性病灶可見於心包,但更常見的是心包腔被纖維性、有時膠凍狀的沾黏所閉塞。心肌膠原蛋白的類纖維蛋白壞死通常見於結締組織隔,偶爾也影響相關的動脈。Libman-Sacks 心內膜炎是 SLE 最著名的心臟表現,據信發生於 30% 至 60% 進行屍檢的病人。小型顆粒狀贅生物已在所有瓣膜表面被發現,雖然二尖瓣與三尖瓣最常受影響。贅生物可能蔓延至侵犯腱索 (chordae tendinae) 與鄰近心內膜。組織學上,它們由覆蓋在退化膠原蛋白上的纖維蛋白組成,並伴隨慢性發炎細胞浸潤。感染性心內膜炎是偶發但重要的併發症。通常可見糖胺聚醣量增加,有時存在血鐵質小體。
肺臟侵犯的組織學病灶包括間質性肺炎、纖維化性肺泡炎與梗塞。在一項大型 SLE 病人屍檢研究中,胸膜肺侵犯在 97.8% 的病人中被偵測到,包括胸膜炎(77.8%)、細菌感染(57.8%)、原發性與繼發性肺泡出血(25.6%)、遠端氣道改變(21.1%)、伺機性感染(14.4%)與血栓栓塞(7.8%)。肺切片的直接免疫螢光檢查可能顯示免疫球蛋白與補體的存在。
淋巴結常顯示反應性增生;偶爾有顯著的病理特徵,可能與淋巴瘤浸潤混淆。這些變化包含壞死性淋巴結炎並伴突出的血鐵質小體。存活的濾泡顯示反應性增生與明顯的漿細胞及免疫母細胞。胸腺可能顯示突出的生發中心形成,並伴 Hassall 小體 (Hassall corpuscles) 的大小與數量增加。脾被膜的脾周炎(「糖霜」‘sugar icing’)常見,一個特徵性的組織學發現是 Malpighian 小體中央(筆毛 penicillary)動脈外膜的同心性纖維化(「洋蔥皮」‘onion skinning’)。
關節表現包括滑膜內的類纖維蛋白變性、類風濕特徵與動脈炎。肝臟可見多種病灶,包括脂肪變性、局部肝細胞壞死與血管炎的證據。中樞神經系統表現有缺血性的致病機轉,最可能基於免疫複合體媒介的血管炎。有些證據指向一種抗神經元自體抗體。新生兒紅斑性狼瘡的心臟病理包含房室結 (atrioventricular node),以及較少程度的竇房結 (sinoatrial node) 之纖維化與鈣化。局部淋巴球性心肌炎與心內膜彈性纖維增生症 (endocardial fibroelastosis) 也可能明顯。
新生兒紅斑性狼瘡 (Neonatal lupus erythematosus)
新生兒疾病皮膚病灶的組織學特徵與免疫螢光型態,與 SCLE 所見者相似。最常被記載的發現包括基底角質形成細胞與附屬器上皮的水樣變性、真皮水腫,以及淺層且偶爾深層的血管周圍與附屬器周圍淋巴組織細胞慢性發炎細胞浸潤。在蕁麻疹樣 SCLE 病灶中,嗜伊紅性球可能很突出。此外,一部分新生兒狼瘡病人顯示嗜中性球皮膚病(Sweet 症候群樣)的組織學特徵,包括組織細胞性變異型(第 15 章)。
鑑別診斷 (Differential Diagnosis)
紅斑性狼瘡必須與其他呈現基底細胞液化變性的皮膚病區分,特別是扁平苔癬與皮膚異色症 (poikiloderma)。紅斑性狼瘡缺乏扁平苔癬的楔形顆粒層增厚 (wedge-shaped hypergranulosis) 與鋸齒狀棘層肥厚 (sawtooth acanthosis)。發炎細胞浸潤典型上為附屬器周圍,而非採取帶狀分布。偶爾,肥厚型紅斑性狼瘡與扁平苔癬顯示相當大的重疊;在這類情況下,陽性狼瘡帶試驗可解決問題。
皮膚異色症,無論是先天性或與皮肌炎(罕見地與 SLE)相關,或作為蕈狀肉芽腫 (mycosis fungoides) 的表現,皆以表皮萎縮與明顯的基底細胞液化變性並伴色素失禁為特徵。上真皮可見斑塊狀的淋巴組織細胞浸潤。真皮乳頭水腫與毛細血管擴張典型上存在。雖然顯然有組織學重疊,但非常不同的臨床表現應可避免任何診斷困難。
非典型淋巴球的存在將可清楚區分與蕈狀肉芽腫相關的變異型。
紅斑性狼瘡的組織學特徵有時難以與多形性日光疹區分,尤其當後者伴有陽性帶試驗時(見上文)。多形性日光疹是最常見的光皮膚病,通常在年輕人,特別是女性,於暴露於紫外線後表現為反覆的紅斑性丘疹、水疱與/或斑塊 (Figs 17.80 and 17.81)。病灶在數小時至數天的潛伏期後發展,常於數天內完全消退,並癒合而無後遺症。組織學上,常可見淺層真皮水腫,以及輕度至中度的淺層與深層血管周圍淋巴組織細胞浸潤。在早期病灶中,輔助誘導型 T 淋巴球 (helper-inducer T lymphocytes) 占多數,且存在數量增加的真皮 Langerhans 細胞。隨著慢性化,細胞毒性抑制型 T 細胞 (cytotoxic-suppressor T cells) 變得更明顯。基底細胞水樣變性通常缺如,且表皮萎縮並非特徵。
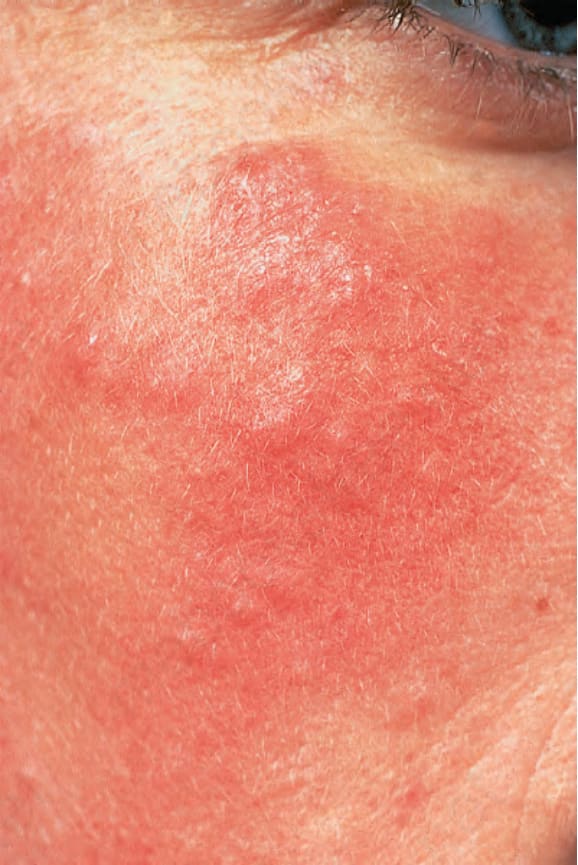
圖 17-80:多形性日光疹——病人在日曬皮膚上表現為紅斑性丘疹與水疱。
Fig. 17.80 Polymorphic light eruption: patients present with erythematous papules and vesicles on sun-exposed skin. By courtesy of the Institute of Dermatology, London, UK.
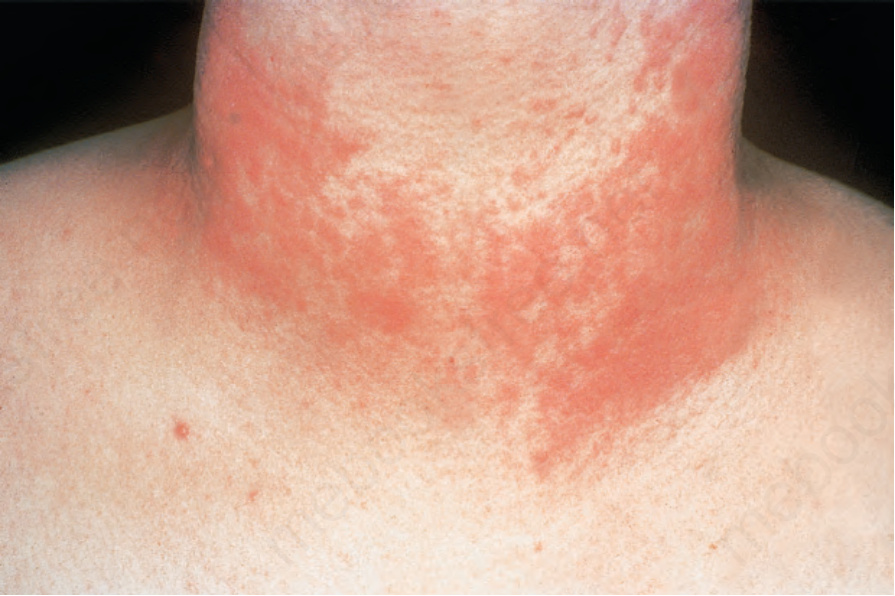
圖 17-81:多形性日光疹——此皮疹典型上對稱,通常會搔癢。
Fig. 17.81 Polymorphic light eruption: the eruption is typically symmetrical and is usually pruritic. By courtesy of the Institute of Dermatology, London, UK.