疾病定義與分類
- 皮下脂膜炎樣 T 細胞淋巴瘤 (Subcutaneous panniculitis-like T-cell lymphoma, SPTCL) 於 1991 年首次描述,為罕見腫瘤,可伴噬血症候群 (hemophagocytic syndrome)。過去許多病例曾被歸為 cytophagic histiocytic panniculitis、Weber-Christian disease、histiocytic lymphoma、malignant histiocytosis 或 histiocytic medullary reticulosis。
- 2001 WHO 分類認定為獨立疾病。其後區分 alpha-beta 與 gamma-delta 譜系:前者多為 CD4–、CD8+、CD56– 表型且預後佳;後者多為侵襲性腫瘤,呈 CD4–、CD8–、CD56+ 表型。現行 EORTC/WHO 分類 (含 2016 更新) 將 SPTCL 一詞保留給 alpha-beta 陽性病例,gamma-delta 病例則視為獨立的「gamma-delta 皮膚 T 細胞淋巴瘤」。
致病機轉
- SPTCL 病因不明。相當比例病例伴自體免疫疾病,尤其紅斑性狼瘡;狼瘡性脂膜炎 (lupus erythematosus panniculitis, LEP) 與 SPTCL 可有臨床與病理重疊,故有人提出兩端為 LEP 與 SPTCL 的疾病譜。基因表現譜研究顯示 SPTCL 上調與自體免疫疾病 (含狼瘡) 相關基因。SPTCL 亦帶有可辨識其為皮膚 T 細胞淋巴瘤中特定亞群的染色體異常。
臨床特徵
- 女性較多 (2:1)。年齡範圍廣,含兒童與老人;高達 20% 病人診斷時 <20 歲,中位年齡 36 歲。
- 表現為紅斑或紫紅色結節或深部斑塊,大小從 <1 至 >20 cm;潰瘍不常見。多數病灶就診時已泛發,侵犯四肢與軀幹,較少及於臉、頸、腋、鼠蹊與臀部。孤立或局限性病灶較少見。
- B 症狀見於 50% 病人;常見貧血、血球減少、ESR 上升與肝功能異常。通常無淋巴結病變;肝脾腫大若出現並非淋巴瘤浸潤所致。
- 噬血症候群見於高達 20% 病例,特徵為肝脾腫大、凝血病變伴廣泛出血、體重減輕、發燒與肌痛。
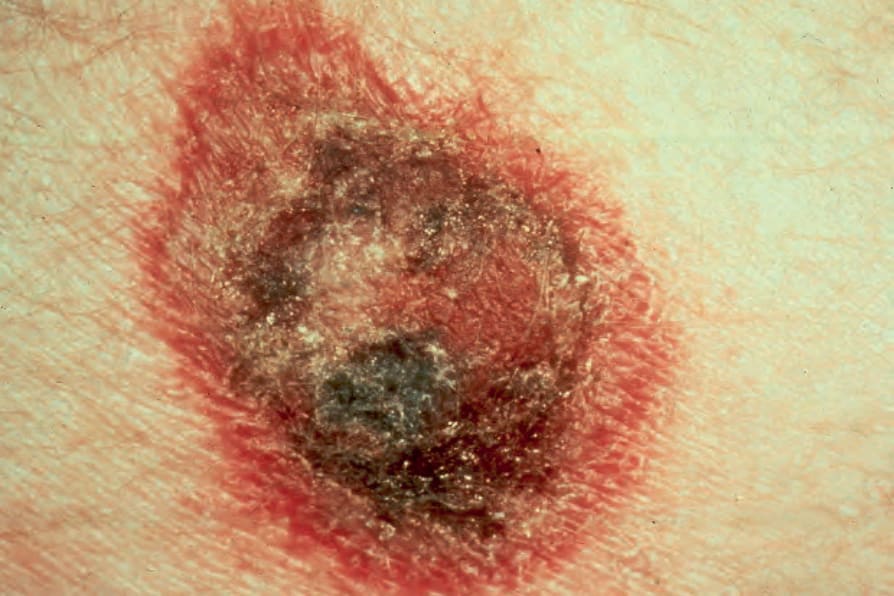
圖 29-151:皮下脂膜炎樣 T 細胞淋巴瘤,此病人雙腿出現多發潰瘍性結節。
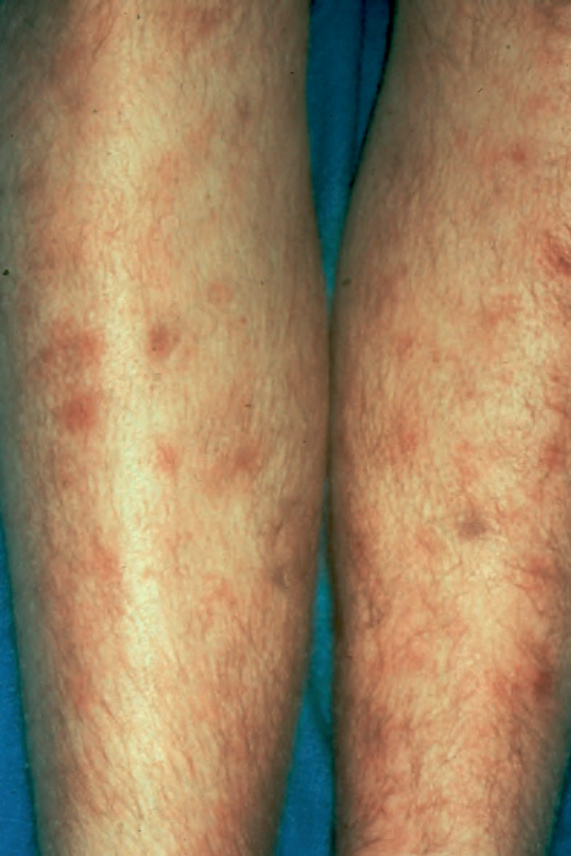
圖 29-152:皮下脂膜炎樣 T 細胞淋巴瘤,此圖病灶類似結節性紅斑 (erythema nodosum)。
組織病理特徵
- 浸潤典型侵犯脂肪小葉 (fat lobules) 而間隔受侵有限;可延伸至深層網狀真皮,但淺層真皮與表皮通常不受累。血管侵犯與血管破壞不常見。
- 腫瘤淋巴球大小不一,為小至中型淋巴球,僅散在大細胞;核深染、胞質稀少。脂肪細胞周圍環繞 (rimming) 為典型,但可為局部且非特異。核碎裂與脂肪壞死幾乎總是出現,常伴組織球,偶見肉芽腫性發炎。組織球常呈空泡狀,可吞噬核碎屑或呈紅血球吞噬 (erythrophagocytosis)。可見反應性小淋巴球;嗜中性球與嗜酸性球罕見。漿細胞與淋巴濾泡稀少,除非與 LEP 重疊。
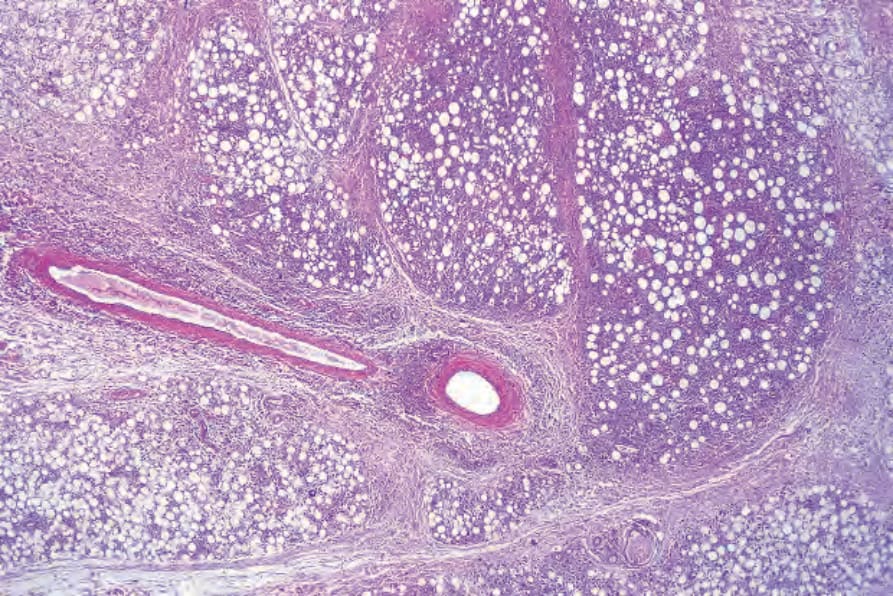
圖 29-153:皮下脂膜炎樣 T 細胞淋巴瘤,皮下脂肪內緻密浸潤,形成特徵性蕾絲狀外觀。
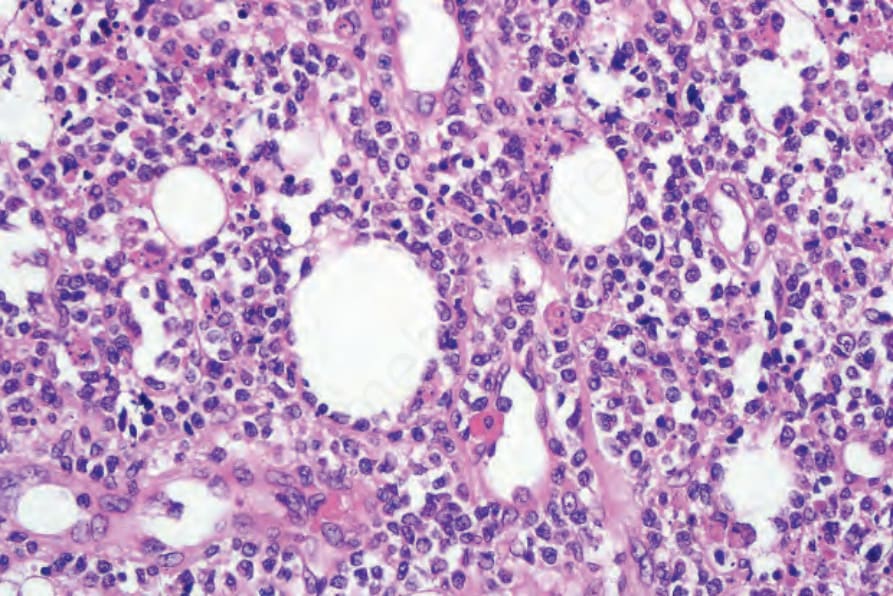
圖 29-154:皮下脂膜炎樣 T 細胞淋巴瘤,腫瘤細胞核可呈囊泡狀或深染,注意特徵性的脂肪細胞環繞。
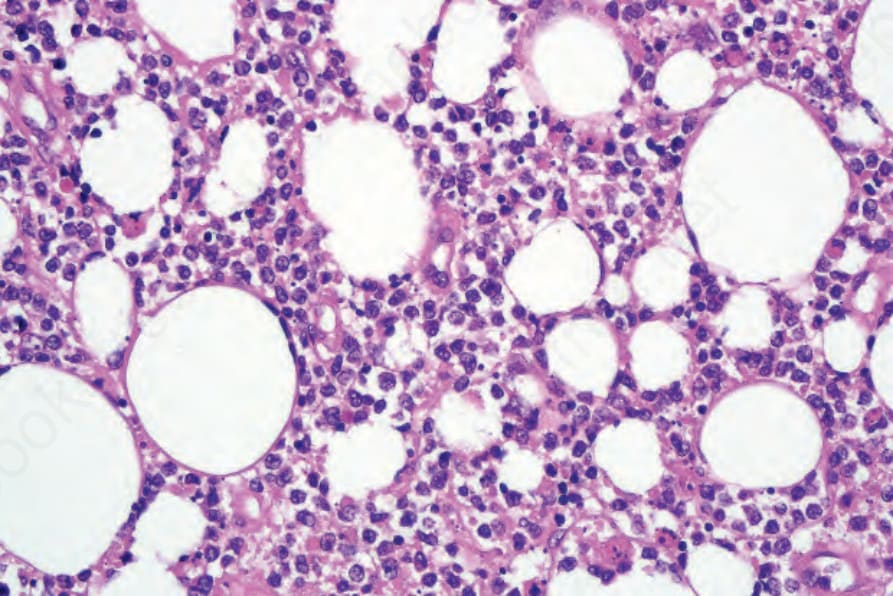
圖 29-155:皮下脂膜炎樣 T 細胞淋巴瘤,腫瘤細胞與組織球混雜。
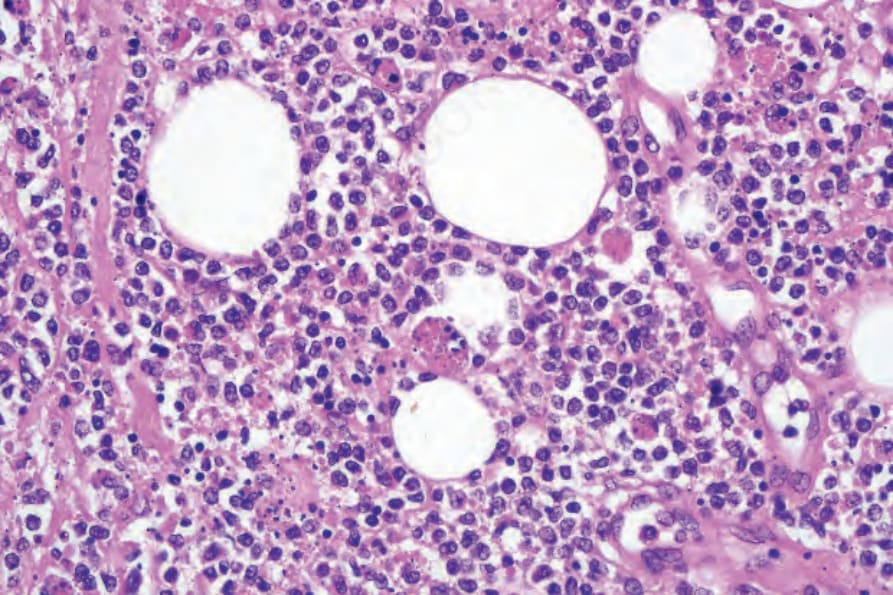
圖 29-156:皮下脂膜炎樣 T 細胞淋巴瘤,此視野顯示明顯的紅血球吞噬。
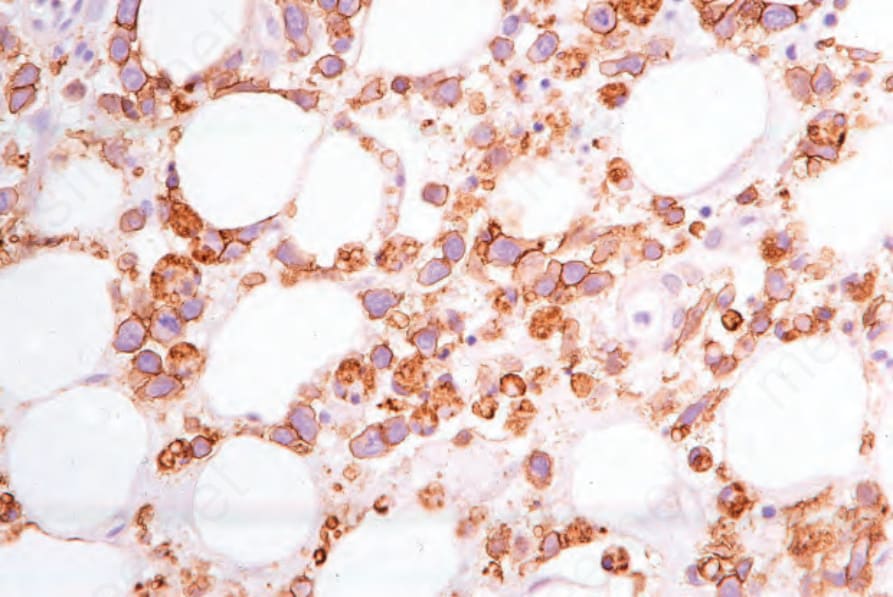
圖 29-157:皮下脂膜炎樣 T 細胞淋巴瘤,淋巴球表現 CD3。
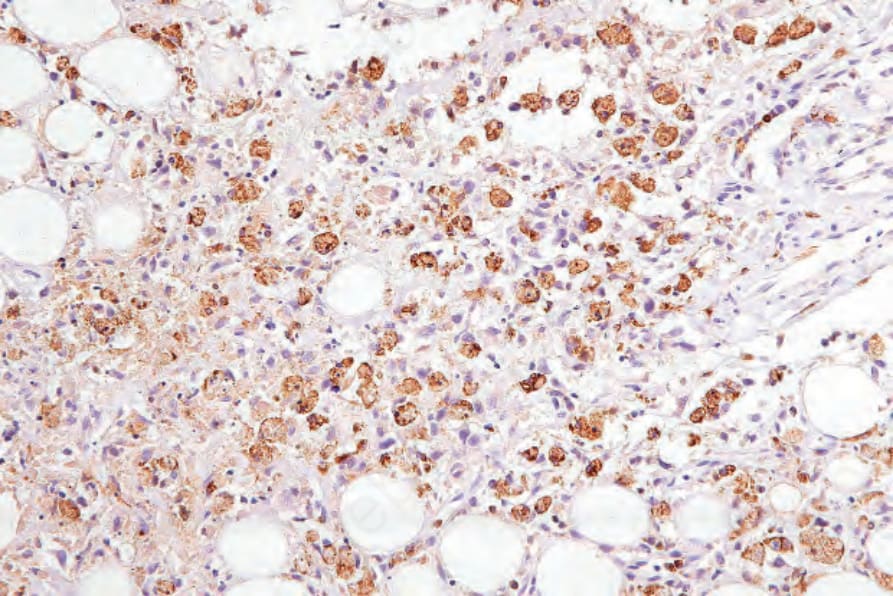
圖 29-158:皮下脂膜炎樣 T 細胞淋巴瘤,組織球由 CD68 標示。
免疫表型與分子檢查
- 腫瘤細胞呈細胞毒表型:CD3+、CD4–、CD8+,並表現細胞毒分子 granzyme B、TIA-1 與 perforin;罕見 CD4+、CD8– 或雙陰性病例。pan-T 抗原喪失:CD2、CD5、CD7 分別喪失於 10%、50%、44% 病例。CD30 通常陰性,罕見少數 CD56+ 細胞。Beta F1 通常可辨識,符合 alpha-beta 表型。EBV 缺如 (亞洲少數例外)。
- 一般可辨識 T 細胞受體基因克隆性重組。
鑑別診斷
- 主要鑑別包括皮膚 gamma-delta T 細胞淋巴瘤與鼻型結外 T/NK 細胞淋巴瘤;免疫組化通常即可區分。皮膚 gamma-delta T 細胞淋巴瘤多為 CD4–、CD8–、缺 beta F1、典型表現 CD56;結外 T/NK 細胞淋巴瘤通常缺 CD3、CD4、CD8 並表現 CD56,且 EBV 染色一律陽性。
- LEP 可能難與 SPTCL 區分;建議篩查所有疑似 SPTCL 病例是否有紅斑性狼瘡。出現大量漿細胞、玻璃樣變區域、反應性生發中心並伴表皮侵犯時,應強烈考慮 LEP;CD123 陽性漿細胞樣樹突細胞聚集亦可見;多株型基因重組亦支持狼瘡診斷。
病程與治療
- SPTCL 通常終其病程局限於皮膚,死亡常因噬血症候群或治療併發症。
- 早期文獻認為 SPTCL 為高死亡率的侵襲性疾病,主要係納入 gamma-delta 陽性病例所致;若用較嚴格標準定義,SPTCL 預後佳,5 年整體與疾病特異性存活率為 80–90%。伴噬血症候群者預後較差,5 年整體存活率僅 46%。
- 雖然 (尤其伴噬血症候群者) 可能需多藥化療,但第一線可用免疫調節劑如類固醇與 ciclosporin A,對部分病人有效。