定義與分類
- 鬆皮症(cutis laxa,廣義 generalized elastolysis)為一組極罕見的彈性組織系統性疾病總稱,含家族型 (familial) 與後天型 (acquired) 變異,後天型常以局部病灶表現。
- 可伴隨多種遺傳/偶發性疾病,如 hereditary gelsolin amyloidosis、Keutel syndrome、geroderma osteodysplastica、Wilson disease、Costello syndrome 等。
- 分類正由歷史性的臨床分型轉向以基因為基礎之系統。藉助遺傳學,目前已知超過 12 種遺傳變異,多數為 autosomal recessive;亦有 autosomal dominant 與 X-linked recessive 變異。Autosomal recessive 型通常較嚴重。
家族(遺傳)型
- 歷史上 type 1 為原型,含皮膚、肺、血管病灶併憩室 (diverticula);少數有甲狀腺低下。
- Autosomal recessive 致病基因包括 FBLN5、EFEMP2、LTBP4、PYCR1、ATP6V0A2、ATP6V1E1、ATP6V1A、ALDH18A1;其中 FBLN5 與 ALDH18A1 與 dominant 型重疊。
- Type 2(Debre type):鬆皮症併骨營養不良、先天性髖關節脫位、智能障礙,少數藍鞏膜;為 ATP6V0A2 突變。
- Type 3(De Barsy syndrome):鬆皮症併早老 (progeria)、角膜混濁、手足徐動、智能障礙併腦發育不良;為 ALDH18A1 突變。
- LTBP4 突變者又稱 Urban-Rifkin-Davis syndrome,可多系統嚴重缺陷。
- Wrinkly skin syndrome 為 recessive 型(type 2, Debre)臨床較輕變異,帶 ATP6V0A2 突變。
- MACS syndrome(巨頭、禿髮、鬆皮症、脊柱側彎)為 RIN2 (20p11.23) recessive 突變。Geroderma osteodysplastica 為 GORAB (1q24.2) recessive 突變。
- X-linked recessive cutis laxa(occipital horn syndrome)為 ATP7A (Xq21.1) 突變,與 Menkes disease 對偶基因相關(銅運輸缺陷);特徵為枕骨角、關節過度伸展、傷口癒合不良、膀胱憩室。
臨床特徵
- 出生時或不久後出現,嬰兒於身體下垂部位有粗糙、鬆弛、冗餘皮膚皺褶;皮膚完全喪失彈性回縮力。
- 顏面侵犯可呈 ‘bloodhound facies’。系統性病灶含肺氣腫、支氣管擴張、血管病變(動脈瘤、瓣膜閉鎖不全)、胃腸憩室與多發疝氣(臍、腹股溝、橫膈)。聲帶受累致聲音變粗變低;外翻 (ectropion) 為典型。
- 遺傳型(尤其 recessive)為嚴重疾病,可因心肺併發症致死。
後天型變異
- 發炎後彈性溶解與鬆皮症(Marshall syndrome):兒童以蕁麻疹樣或環狀紅斑丘疹疹合併彈性組織破壞及後續皮膚鬆弛;Sweet syndrome 可誘發。
- 成人全身性後天鬆皮症:較先天型少見,常伴肺與心血管侵犯及憩室;可原發或繼發於多種皮膚病,並可併 penicillamine、isoniazid、penicillin 敏感、multiple myeloma、單株免疫球蛋白病等。
- 局部性鬆皮症:限於單一部位,可繼發於梅毒、結節病、靜脈曲張等。
致病機轉
- 屬廣義 elastin 缺陷疾病譜(含 Marfan、Loeys-Dietz、pseudoxanthoma elasticum、部分 Ehler-Danlos 等)。各病皆有影響彈性纖維結構/生成/處理/穩定的特定基因缺陷。
- Elastin 約占成熟彈性纖維的 90%。ELN 突變可致 elastin 量性與功能性缺陷,單一等位基因突變即可為 dominant negative 致病。
- Fibulin-5 (FBLN5) 引導 elastin 與微纖維作用;fibulin-4 (EFEMP2) 與 LOX 編碼之 lysyl oxidase 協同進行 elastin 交聯。LTBP4 缺陷致微纖維增厚波浪化、功能受損。
- ATP6V0A2 等 H+-ATPase 缺陷影響囊泡酸化、運輸與分泌(部分透過醣化改變)。ALDH18A1 與 PYCR1 為粒線體代謝酶,突變失調氧化代謝及 ornithine/proline 生合成,可解釋更廣的早老與神經退化效應。
- 後天型機轉不確定,可能與局部 elastase 過度生成有關(尤其發炎後病人);部分病人帶 FBLN5/ELN 雜合缺陷,提示對發炎後損傷之易感性。
組織病理
- 以彈性纖維數目減少及相關退化變化為特徵。纖維可於乳頭層、網狀層或兩者皆缺;網狀層內者常縮短、變尖、退化。
- 發炎後型可見大量真皮急性發炎細胞浸潤(含嗜中性球與嗜酸性球),並描述嗜中性球圍繞彈性纖維呈柵欄狀;有時見血管炎與肉芽腫性皮膚炎。
- H&E 切片組織學常無顯著異常,僅見輕度血管周圍淋巴球浸潤。
- 超微結構:病灶纖維碎裂、分支、電子緻密、退化,但無 pseudoxanthoma elasticum 所見之鈣化;可見組織球進行彈性吞噬 (elastophagocytosis)。
- 免疫組化或特殊染色可確認部分型別之 elastin 喪失;分子診斷(單一或基因組套)日益用於確認遺傳病例並供遺傳諮詢。
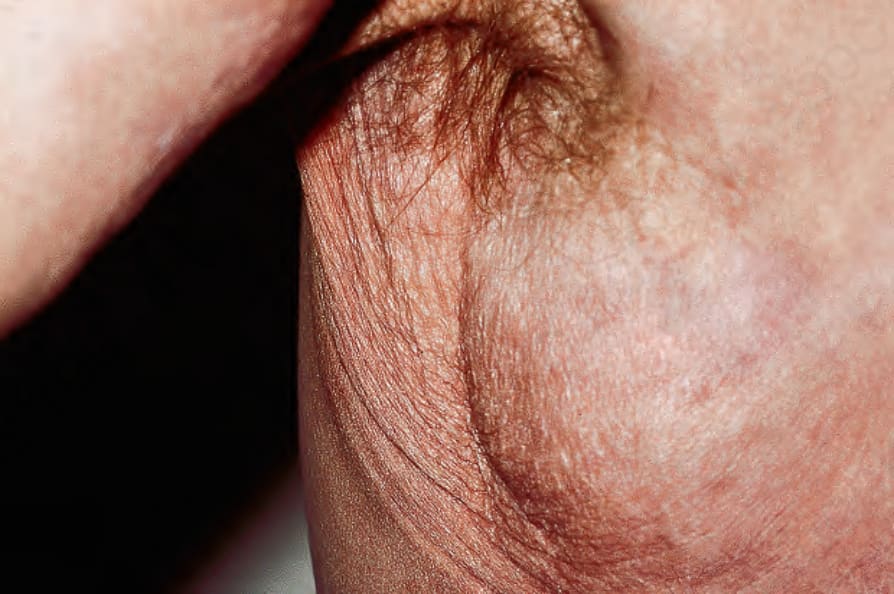
圖 21-40:鬆皮症,腋窩可見明顯皮膚皺褶;此為併發皮膚類澱粉沉積導致彈性層破壞之極不尋常後天型。

圖 21-42:鬆皮症,此嬰兒皮膚極鬆弛,拉伸放開後不回復原位,並注意臍疝。
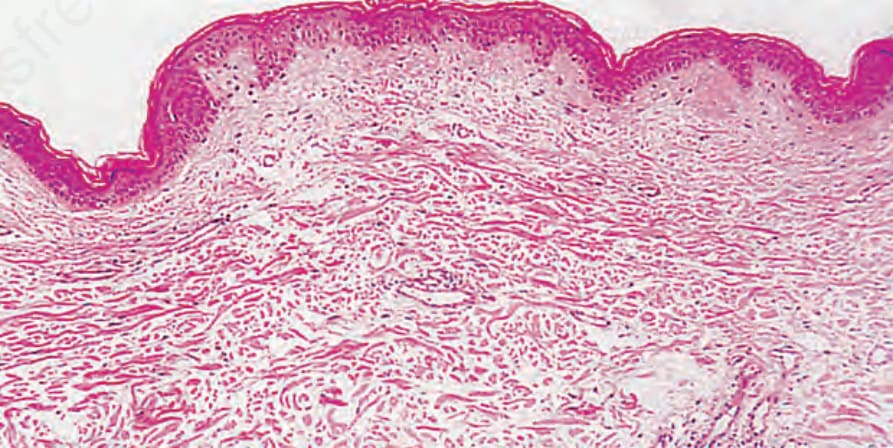
圖 21-43:鬆皮症,H&E 切片組織學相當不顯著,僅見輕度血管周圍淋巴球浸潤。
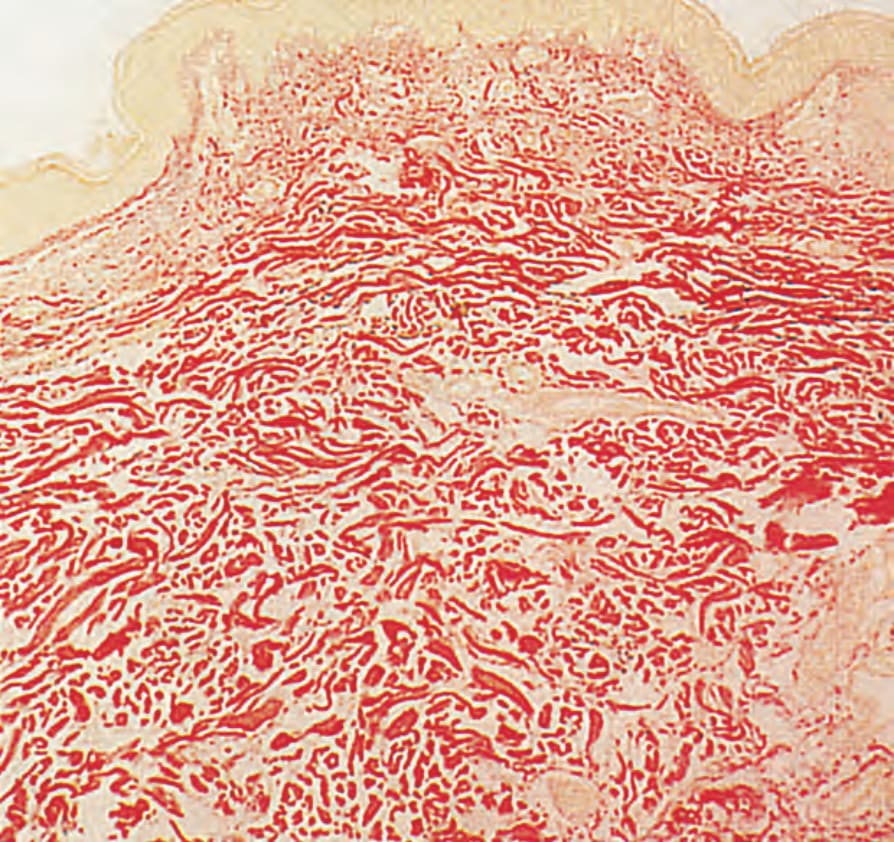
圖 21-44:鬆皮症,網狀真皮彈性組織廣泛喪失,乳頭層相對保留。